|
© Borgis - Postępy Nauk Medycznych 12, s. 912-980
Dr n. med. Wojciech Bik
Ocena zależności wydzielania adipokin i insulinooporności oraz asocjacji wybranych polimorfizmów genów adiponektyny i rezystyny w otyłości
Assessment of correlations between adipokine release and insulin resistance, and evaluation of selected polymorphisms of adiponectin and resistin genes in obesity
Centrum Medyczne Kształcenia Podyplomowego w Warszawie
Streszczenie
Słowa kluczowe: otyłość, zespół metaboliczny, adipokiny, polimorfizmy genów adiponektyny i rezystyny
Summary
Key words: obesity, metabolic syndrome, adipokines, polymorphisms of adiponectin and resistin genes
Wstęp
Otyłość jest przewlekłą chorobą metaboliczną wynikającą z zaburzeń homeostazy energetycznej organizmu, która prowadzi do zwiększenia ilości tkanki tłuszczowej, a w konsekwencji do rozwoju powikłań wielonarządowych (w tym chorób układu sercowo-naczyniowego, kostno-stawowego, zaburzeń gospodarki lipidowej).
Nadwaga i otyłość stanowią istotny problem epidemiologiczny i społeczny ze względu na dynamiczny wzrost częstości ich występowania, przede wszystkim w krajach wysokorozwiniętych.
Zespół metaboliczny
Wieloletnie obserwacje kliniczne osób otyłych doprowadziły do wyodrębnienia i zdefiniowania pod koniec lat osiemdziesiątych XX wieku zespołu metabolicznego, którego patogenetycznym podłożem jest hiperinsulinizm z insulinoopornością. Zespół ten skojarzony jest z otyłością trzewną, dyslipidemią, nadciśnieniem tętniczym i zaburzeniami gospodarki węglowodanowej. Zespół metaboliczny zwiększa ponad 3-krotnie ryzyko cukrzycy typu 2 oraz chorób układu sercowo-naczyniowego.
Tkanka tłuszczowa jako narząd wydzielania wewnętrznego
Od momentu odkrycia w 1994 roku przez Friedmana i wsp. leptyny oraz jej receptorów, tkanka tłuszczowa przestała być uważana jedynie za rezerwuar energetyczny ustroju stała się obiektem wnikliwych badań, które w konsekwencji doprowadziły do odkrycia licznych adipokin (substancji wydzielanych przez adipocyty), jak również wykazały istotną aktywność metaboliczną komórek tkanki tłuszczowej.
Do najważniejszych adipokin zaliczamy: adiponektynę i jej frakcję wysokocząsteczkową (HMW adiponektyna), rezystynę, wisfatynę i leptynę.
Adiponektyna
Adiponektyna jest polipeptydem o masie cząsteczkowej 33 kDa syntetyzowanym przez tkankę tłuszczową. Stężenie adiponektyny wzrasta wraz z redukcją masy ciała Natomiast wraz ze wzrostem BMI stwierdzany jest spadek adiponektyny w surowicy krwi obwodowej.
Niezwykle istotnym metabolicznie działaniem adiponektyny jest jej wpływ na zmniejszenie insulinooporności Poza korzystnym działaniem antydiabetogennym oraz przeciwzapalnym, peptyd ten może wykazywać działanie przeciwmiażdżycowe. Obniżenie stężenia adiponektyny stwierdzono u pacjentów z chorobą niedokrwienną serca.
Adiponektyna w surowicy może występować w postaci kilku frakcji, w tym: dimerów, trimerów, heksamerów lub multimetów. Frakcja wysokocząsteczkowa adiponektyny (tzw. HMW adiponectin), składająca się z ok. 16 cząsteczek tego peptydu, wydaje się być najbardziej aktywną formą tej adipokiny. Wzrost poziomu HMW adiponektyny lub stosunku HMW adiponektyny do adiponektyny całkowitej koreluje dodatnio ze wzrostem insulinowrażliwości.
Jednakże rola i znaczenie adiponektyny w wielu procesach patologicznych nie są do końca jednoznacznie wyjaśnione. Wyniki niektórych badań wskazują, że wyższe poziomy całkowitej adiponektyny mogą być negatywnym czynnikiem prognostycznym u osób z chorobami układu sercowo-naczyniowego, a także w przewlekłej niewydolności krążenia.
Wydaje się, że działanie adiponektyny jest niezwykle korzystne z metabolicznego punktu widzenia, jednakże istnieją kontrowersje co do jej roli w stanach patologicznych i zatem aspekt ten wymaga dalszej wnikliwej analizy.
Rezystyna
Rezystyna jest białkową adipokiną syntetyzowaną w adipocytach, ale jej ekspresję stwierdzono także w komórkach jednojądrowych krwi obwodowej Podwyższone wartości rezystyny wykazano w zwierzęcym modelu otyłości. Wydaje się, że nasilanie insulinooporności przez rezystynę spowodowane jest stymulacją glukoneogenezy i glikogenolizy. Rezystyna ma również wpływ na pogorszenie utylizacji glukozy w mięśniach szkieletowych.
Wisfatyna
Wisfatyna jest polipeptydem o działaniu insulinomimetycznym produkowanym przede wszystkim przez trzewną tkankę tłuszczową.
Podwyższone wartości wisfatyny stwierdzane są w otyłości, a także w insulinooporności. Ponadto wyższe poziomy wisfatyny wykazano u chorych z przewlekłymi chorobami nerek, u których wisfatyna może nasilać dysfunkcję śródbłonka naczyniowego. Rola, jak również mechanizm działania wisfatyny nie zostały jeszcze w pełni wyjaśnione.
Leptyna
Leptyna syntetyzowana jest przede wszystkim w adipocytach białej tkanki tłuszczowej oraz w niewielkich ilościach w brunatnej tkance tłuszczowej.
Podstawową funkcją leptyny jest kontrola bilansu energetycznego ustroju w mechanizmie hamowania łaknienia na poziomie podwzgórza. Poposiłkowy wzrost stężenia leptyny powoduje obniżenie poziomu neuropeptydu Y (NPY).
U osób otyłych stwierdzany jest często podwyższony poziom leptyny, spowodowany leptynoopornością.
Publikacje dotyczące wpływu leptyny na procesy rozwoju miażdżycy wskazują, że leptyna wywołuje wielorakie efekty aterogenne, m.in. wpływając na dysfunkcję śródbłonka, stymulując stres oksydacyjny i reakcję zapalną, zwiększając agregację płytek krwi oraz proliferację komórek mięśniówki gładkiej naczyń.
Rola neuropeptydów wydzielanych w ośrodkowym układzie nerwowym i obwodowo w kontroli łaknienia
Zaburzenie regulacji łaknienia stanowi istotny czynnik wpływający na rozwój nadwagi i otyłości. Szeroko pojęte zachowania żywieniowe kontrolowane są przez neuropeptydy wydzielane w ośrodkowym układzie nerwowym i przewodzie pokarmowym, adipokiny produkowane w tkance tłuszczowej. Istotne znaczenie w kontroli ilości i jakości przyjmowanych pokarmów odgrywa podwzgórze, w którym zlokalizowane są ośrodki głodu i sytości.
Neuropeptydy, zarówno pochodzenia centralnego jak i obwodowego, zostały podzielone na dwie przeciwstawne grupy: peptydy oreksygeniczne i anoreksygeniczne. Najważniejszymi peptydami oreksygenicznymi są syntetyzowane w OUN: neuropeptyd Y, oreksyny, Agouti-related peptide (AgRP), galanina, endogenne opioidy, peptydy układu endokanabinoidowego oraz syntetyzowana, przede wszystkim w komórkach żołądka, grelina. Do drugiej grupy, peptydów anoreksygenicznych, zalicza się: peptydy ośrodkowe, w tym cocaine-amphetamine regulated transcript (CART), proopiomelanokortynę (POMC), czynnik pobudzający wydzielanie kortykotropiny (CRF), serotoninę, a do wydzielanych w przewodzie pokarmowym cholecystokininę, peptyd YY, amylinę, insulinę i bombezynę oraz syntetyzowaną w tkance tłuszczowej leptynę.
Peptydy obwodowe
Poza neuropeptydami wywierającymi swoje działanie na poziomie centralnego układu nerwowego istotną rolę odgrywają peptydy syntetyzowane w tkankach obwodowych, przede wszystkim w przewodzie pokarmowym, do których zaliczamy grelinę (peptyd oreksygeniczny) i cholecystokininę (peptyd anoreksygeniczny).
Grelina jest 28-aminokwasowym peptydem wyizolowanym początkowo z żołądka szczura. Peptyd ten syntetyzowany jest nie tylko w komórkach śluzówki żołądka, ale także w przysadce, podwzgórzu, trzustce, płucach oraz układzie immunologicznym. Grelina jest hormonem przewodu pokarmowego, którego poziom wzrasta w czasie głodzenia, co jest bodźcem stymulującym łaknienie.
Zaburzenia hormonalne w otyłości
Wieloletnie obserwacje i badania potwierdzają, że układ hormonalny włączony jest w skomplikowany system kontroli łaknienia i homeostazy energetycznej ustroju.
Wśród licznych hormonów, mających wpływ na kontrolę łaknienia jest insulina. Insulina nasila oksydację glukozy, magazynowanie lipidów i aminokwasów, natomiast hamuje lipolizę, oksydację tłuszczów oraz uwalnianie glukozy zmagazynowanej w postaci glikogenu. Cechą charakterystyczną dużej grupy pacjentów z nadwagą, a przede wszystkim z otyłością jest insulinooporność często połączona z hiperinsulinizmem. Redukcja masy ciała uzyskiwana przy pomocy samej diety, jak też w połączeniu z farmakoterapią lub leczeniem operacyjnym prowadzi nie tylko do zmniejszenia ilości tkanki tłuszczowej i redukcji masy ciała ale także do zmniejszenia insulinooporności.
Poza insuliną istotną rolę w rozwoju otyłości oraz zaburzeń metabolicznych mogą ogrywać także hormony tarczycowe, hormon wzrostu oraz hormony kory nadnerczy.
Czynniki genetyczne otyłości
Otyłość związana jest z wieloma czynnikami genetycznymi i środowiskowymi. Badania rodzin osób otyłych wykazują, że czynniki genetyczne wpływają na BMI w 30-40%, natomiast oddziaływania czynników środowiskowych wynoszą ok. 60-70%.
Czynniki genetyczne bez wątpienia warunkują predyspozycje do wystąpienia otyłości, jednakże określanie otyłości jako choroby uwarunkowanej genetycznie wydaje się mieć ograniczone uzasadnienie. Pod względem uwarunkowań genetycznych z otyłości można wydzielić choroby dziedziczone jedno- lub wielogenowo. W patogenezie otyłości najlepiej poznanymi mutacjami pojedynczego genu są mutacje: genu receptora melanokortyny 4, genu leptyny lub genu receptora leptyny. Wydaje się, że dziedziczenie wielogenowe ma zasadnicze znaczenie w występowaniu predyspozycji do otyłości.
W ostatnim czasie opublikowano szereg doniesień dotyczących polimorfizmu genu FTO (FAT mass and obesity-associated gene) jako istotnego czynnika w rozwoju otyłości. Badania prowadzone w Europie wskazują, że wśród dzieci i młodzieży obecność niektórych polimorfizmów tego genu prowadzić może do wzrostu masy ciała związanego z większą ilością przyjmowanych pokarmów i słabiej wyrażonym poposiłkowym uczuciem sytości. Wyniki badań dotyczące genu FTO są niejednoznaczne i wskazują na konieczność prowadzenia dalszych badań w tym zakresie w celu uzyskania bardziej jednoznacznych wyników.
Kolejnym istotnym czynnikiem genetycznym, który wydaje się także mieć znaczenie w rozwoju otyłości jest gen receptora aktywowanego proliferatorem peroksysomów kodujący receptor PPARγ. Badania polimorfizmów genu tego receptora u ludzi wykazały, że występowanie polimorfizmu pro12ala może wiązać się ze zmniejszonym ryzykiem cukrzycy typu 2, zwiększoną wrażliwością na insulinę i niższym BMI.
Zaburzenia genetyczne mogą być także związane z syntezą lub działaniem adipokin. Wśród potencjalnie istotnych zaburzeń rolę odgrywają polimorfizmy związane z genem adiponektyny. Są to zmiany pojedynczego nukleotydu w regionie promotorowym, zmiany w genie powodujące przyłączenie glicyny w pozycji 15 polipeptydu tj. w sekwencji sygnałowej, która to sekwencja nie występuje w dojrzałym białku (SNP 45 (T/G). Polimorfizmy mogą też dotyczyć zmian w obrębie intronów lub zmian w obrębie tzw. domeny włóknistej lub globularnej cząsteczki adiponektyny. Wymienione powyżej zmiany o podłożu genetycznym związane mogą być z większym prawdopodobieństwem wystąpienia cukrzycy typu 2, insulinooporności oraz nadciśnienia tętniczego. Uznaje się, że omówione uprzednio polimorfzmy genu adiponektyny mogą także zwiększać ryzyko wystąpienia zespołu metabolicznego.
Rezystyna jest adipokiną związaną prawdopodobnie z rozwojem insulinooporności w przebiegu otyłości i zespołu metabolicznego. Wyższe stężenia rezystyny w osoczu i surowicy stwierdzono u chorych z cukrzycą i otyłością. Do najczęściej opisywanych w literaturze polimorfizmów rezystyny należy polimorfizm -420C/G zlokalizowany w części promotorowej genu. Polimorfizm ten związany jest z występowaniem otyłości, insulinooporności i cukrzycy typu 2 w różnych badanych populacjach.
Wyniki badań dotyczących związku polimorfizmów genu rezystyny z występowaniem cukrzycy typu 2, otyłości, nadciśnienia tętniczego a w konsekwencji zespołu metabolicznego są niejednoznaczne i wymagają dalszych szczegółowych badań.
Uzasadnienie celowości podjęcia badań
Dotychczas opublikowano szereg danych na temat otyłości i zespołu metabolicznego, pochodzących z licznych badań eksperymentalnych i klinicznych ale pomimo to nadal istnieje wiele niejasności dotyczących patofizjologii, jak i skutków zdrowotnych związanych z otyłością i zespołem metabolicznym.
Wraz z rozwojem stopnia zaawansowania klinicznego otyłości mierzonego przy pomocy BMI stwierdzany jest stopniowy wzrost częstości występowania zespołu metabolicznego. Jednakże nawet u pacjentów z otyłością olbrzymią (BMI ≥ 40 kg/m2) zespół metaboliczny nie jest rozpoznany u wszystkich chorych. Od kilku lat w publikacjach ta szczególna grupa chorych bez zespołu metabolicznego określana jest jako otyli metabolicznie zdrowi. Jeżeli zatem pomimo istnienia tak zaawansowanej klinicznie otyłości, pacjenci ci nie mają nasilonych zaburzeń metabolicznych czy też nie stwierdza się u nich cukrzycy typu 2 lub nadciśnienia tętniczego, to istotnym wydaje się być pytanie czy pomiędzy pacjentami otyłymi z lub bez zespołu metabolicznego istnieją różnice w wydzielaniu adipokin oraz czy ewentualnie tym zmianom towarzyszą asocjacje genetyczne. Dotychczas u tych pacjentów nie wykazano jednoznacznych zmian w profilu wydzielanych adipokin lub też odmienności w zakresie wskaźników genetycznych, mających związek z wystąpieniem lub brakiem obecności zespołu metabolicznego.
W niniejszej pracy została podjęta próba oceny czynników genetycznych z zastosowaniem analizy częstości występowania polimorfizmów genów dwóch adipokin o przeciwstawnych działaniach: adiponektyny o działaniu zmniejszającym insulinoooporność, posiadającej właściwości przeciwmiażdżycowe i antydiabetogenne oraz rezystyny, która uznawana jest za adipokinę stymulującą rozwój insulinooporności. Wykazanie istotnych statystycznie różnic w występowaniu poszczególnych genotypów badanych polimorfzmów lub ich asocjacji z zespołem metabolicznym mogłoby stać się potencjalnym predyktorem ewentualnych zaburzeń metabolicznych w otyłości.
Połączenie badań dotyczących zmian stężenia adipokin w powiązaniu z oceną insulinooporności wraz z badaniami polimorfizmów genów adiponektyny i rezystyny może pozwolić na wyodrębnienie grupy pacjentek o szczególnie nasilonych zmianach metabolicznych, które to pacjentki powinny być poddane intensywnemu nadzorowi medycznemu.
Cel pracy
Celem pracy jest wyodrębnienie czynników wpływających na wystąpienie zespołu metabolicznego u osób z nadwagą i otyłością.
Analizie zostaną poddane zmiany stężeń adipokin i peptydów, związanych z kontrolą łaknienia w grupach pacjentek z obecnością lub brakiem zespołu metabolicznego z różnym stopniem zaawansowania otyłości i nadwagi.
Oceniane będą także korelacje pomiędzy analizowanymi peptydami a wskaźnikami isnulinooporności, wynikami badań parametrów lipidowych i parametrami antropometrycznymi.
Badane będą także asocjacje polimorfizmów genów adiponektyny i rezystyny w odniesieniu do obecności zespołu metabolicznego u pacjentek z nadwagą lub otyłością.
Na podstawie przedstawionych powyżej założeń badawczych podjęta zostanie próba określenia ewentualnych markerów peptydowych lub genetycznych, pozwalających na wyodrębnię grupy pacjentek o podwyższonym ryzyku wystąpienia zespołu metabolicznego.
Zadania badawcze
1) Ocena parametrów antropometrycznych, wskaźników gospodarki lipidowej i węglowodanowej w wybranej grupie kobiet oraz kwalifikacja ich do poszczególnych grup badanych:
a) pacjentki szczupłe, zdrowe z BMI <25 kg/m2,
b) pacjentki z nadwagą i otyłością z BMI 25-39,9 kg/m2 skojarzoną z zespołem metabolicznym,
c) pacjentki z nadwagą i otyłością z BMI 25-39,9 kg/m2 bez zespołu metabolicznego,
d) pacjentki z otyłością olbrzymią z BMI ≥ 40 kg/m2.
2) Ocena zmian stężeń adipokin i peptydów (leptyny, adiponektyny całkowitej i wysokocząsteczkowej, wisfatyny, greliny, rezystyny) w poszczególnych badanych grupach pacjentek z nadwagą i otyłością w stosunku do grupy kontrolnej, jak również porównanie pomiędzy poszczególnymi wymienionymi powyżej badanymi grupami pacjentek.
3) Ocena zmian stężeń peptydów, adipokin i wyników badań biochemicznych pomiędzy pacjentkami z obecnością lub brakiem zespołu metabolicznego.
4) Ocena korelacji i zależności zmian stężeń adipokin w stosunku do wskaźników antropometrycznych, gospodarki lipidowej i węglowodanowej w poszczególnych badanych grupach kobiet z nadwagą i otyłością.
5) Ocena zmian częstości występowania badanych polimorfizmów genów adiponektyny i rezystyny oraz ocena ilorazu szans pomiędzy następującymi grupami grupami kobiet:
a) pacjentki z nadwagą i otyłością skojarzoną z zespołem metabolicznym w stosunku do kobiet z nadwagą i otyłością bez zespołu metabolicznego,
b) pacjentki z nadwagą i otyłością skojarzoną z zespołem metabolicznym w stosunku do kobiet z prawidłowym BMI,
c) pacjentki z nadwagą i otyłością bez zespołu metabolicznego w stosunku do kobiet z prawidłowym BMI.
6) Próba określenia asocjacji pomiędzy zespołem metabolicznym a analizowanymi polimorfizmami.
7) Próba znalezienia markerów peptydowych i/lub genetycznych, różnicujących nadwagę i otyłość skojarzoną z zespołem metabolicznym od nadwagi i otyłości bez zespołu metabolicznego.
Materiał i metody
Badania przeprowadzono łącznie u 369 kobiet, które podzielono na następujące grupy: 34 kobiety z otyłością olbrzymią (BMI ≥ 40 kg/m2), 100 kobiet z nadwagą i otyłością i zespołem metabolicznym (BMI 25-39,9 kg/m2), 131 kobiet z nadwagą i otyłością (BMI 25-39,9 kg/m2) bez zespołu metabolicznego. Grupę kontrolną stanowiły 104 młode kobiety z prawidłową masą ciała (BMI 18-24,9 kg/m2).
Zespół metaboliczny rozpoznawano na podstawie kryteriów sformułowanych przez International Diabetic Federation (IDF 2005).
Nadciśnienie tętnicze definiowano wg kryteriów ESH i ESC, natomiast cukrzycę typu 2 rozpoznawano zgodnie z następującymi kryteriami: w przypadku glikemii na czczo ≥ 126 mg/dl lub gdy 2 godziny po podaniu 75 g glukozy glikemia ≥ 200 mg/dl lub kiedykolwiek w ciągu doby glikemia wynosiła ≥ 200 mg/dl w osoczu krwi żylnej. Klasyfikację cukrzycy oceniano wg zaleceń Komitetu Ekspertów WHO sformułowanych w 1998 r.
Kryteria wykluczające z włączenia do badań były następujące:
1. choroby układu endokrynnego,
2. przewlekłe choroby układu oddechowego,
3. przewlekłe choroby wątroby i nerek,
4. choroby nowotworowe.
Żadna z badanych kobiet w trakcie przeprowadzonych badań nie wykazywała objawów ostrej choroby zakaźnej jak również nie otrzymywała leków przeciwzapalnych, nie nadużywała alkoholu i nie paliła papierosów.
Protokół badań został zaakceptowany przez Komisję Bioetyczną w Centrum Medycznym Kształcenia Podyplomowego. Wszystkie osoby badane wyraziły świadomą zgodę na uczestniczenie w badaniu klinicznym i pobranie krwi.
Badanie kliniczne
Pomiar ciśnienia tętniczego, wzrost i wagę oraz BMI oceniano wg standardowych procedur. Obwód talii i wskaźnik talia-biodra (waist-hip ratio WHR) stosowano jako wskaźnik otyłości centralnej. Zawartość tkanki tłuszczowej oceniano metodą elektrycznej bioimpedancji (BIA). Insulinooporność mierzono przy pomocy wskaźnika HOMA-IR
Pomiar stężenia peptydów i hormonów w osoczu krwi obwodowej oraz ocena parametrów lipidowych i gospodarki węglowodanowej
Krew do badań pobierano rano, na czczo po 12 godzinach od ostatniego posiłku. Bezpośrednio po pobraniu materiału, próbki krwi wirowano w temperaturze +4°C, a uzyskaną surowicę przechowywano w temperaturze -70°C.
Profil lipidowy oraz oznaczenia poziomu glukozy wykonywano wg standardowych procedur laboratoryjnych.
Stężenia leptyny, adiponektyny i greliny (active) wykonano metodą radioimmunologiczną (RIA).
Poziomy rezystyny i rozpuszczalnego receptora dla leptyny oceniano, stosując testy ELISA.
Oznaczenia stężenia C-końcowego fragmentu wisfatyny oraz frakcji wysokocząsteczkowej adiponektyny (HMW adiponektyna) wykonano testami metodą EIA.
Insulinę i oznaczano metodą IRMA.
Zmienność wewnątrzseryjna i międzyseryjna wszystkich stosowanych do oznaczeń zestawów była mniejsza niż 10%.
Badania genetyczne – oznaczanie polimorfizmów pojedynczych nukleotydów dla genu rezystyny i adiponektyny
Genomowe DNA było izolowane z krwi pełnej z dodatkiem EDTA przy pomocy zestawu NucleoSpin Blood Quick Pure kit (Macherey-Nagel – Niemcy) według załączonego do zestawu protokołu.
Oznaczano następujące polimorfizmy pojedynczego nukleotydu (SNP) dla genu rezystyny: 420 C/G, 62 G/A, 537 A/C oraz następujące SNP dla genu adiponektyny: 45T/G, 276 G/T, 11 377 C/G, 11 391 G/A. Genotypowanie wykonano przy pomocy TaqMan SNP Genotyping Assays. Reakcja PCR czasu rzeczywistego (Real Time PCR – RT-PCR) była wykonana aparatem ABI PRISM Sequence Detection System (SDS). Rozdział poszczególnych alleli w zakresie kolejnych polimorfizmów wykonywano aparatem ABI PRISM 7000 SDS Software.
Analiza statystyczna
Analizę statystyczną wykonano przy użyciu pakietu STATISTICA ver 7.1 PL, ustalając poziom istotności statystycznej α = 0,05.
Normalność rozkładu w obrębie poszczególnych grup badano testami: Shapiro-Wilka oraz Kołmogorowa-Smirnowa z poprawką Lillieforsa.
Ze względu na brak normalności rozkładu danych w części badanych parametrów do weryfikacji hipotezy, dotyczącej zróżnicowania więcej niż 2 grup, stosowano test Kruskala-Wallisa, a przy porównaniu dwóch grup stosowano test Manna-Whitneya. Korelację pomiędzy badanymi adipokinami oraz wynikami badań antropometrycznych i badań biochemicznych oceniano przy użyciu korelacji Spearmana.
Po zbadaniu założeń wykonano analizę kowariancji ANCOVA w badanych grupach, gdzie jako zmienną zależną analizowano adiponektynę całkowitą i wysokocząsteczkową, leptynę, rezystynę, wisfatynę i grelinę, a jako zmienną wikłającą uznano kolejno: BMI, BIA i WHR.
Wyniki badań peptydów, adipokin, wskaźników antropometrycznych i lipidowych (dane ilościowe) zostały przedstawione jako średnia ± odchylenie standardowe (SD).
Porównania częstotliwości dystrybucji genotypów poszczególnych analizowanych polimorfizmów genów adiponektyny i rezystny wykonano odpowiednio przy pomocy testu χ2.
Obliczono także iloraz szans (OR) wraz z 95% przedziałem ufności (CI). Ocenę tę przeprowadzono dla modelu dominującego, który definiowano jako heterozygoty + homozygoty recesywne vs homozygoty dominujące.
Oceniano również zgodność rozkładu genotypów poszczególnych badanych polimorfizmów z prawem Hardyego-Wienberga w grupie kontrolnej.
W celu oceny asocjacji analizowanych polimorfizmów genów adiponektyny i rezystyny z nadwagą, otyłością i zespołem metabolicznym oraz jednoczesnej oceny ilorazu szans pomiędzy poszczególnymi grupami pacjentek wykonano analizę regresji logistycznej w następujących układach:
1. Pacjentki z otyłością i nadwagą skojarzoną z zespołem metabolicznym w odniesieniu do kobiet z nadwagą i otyłością bez towarzyszącego zespołu metabolicznego.
2. Pacjentki z nadwagą i otyłością z zespołem metabolicznym w stosunku do grupy kobiet młodych z prawidłowym BMI.
3. Pacjentki z nadwagą i otyłością bez zespołu metabolicznego w porównaniu do grupy młodych kobiet z prawidłowym BMI.
Podsumowanie uzyskanych wyników
W wyniku przeprowadzonych badań stwierdzono:
1. Kobiety z otyłością olbrzymią miały najbardziej nasilone zmiany w profilu wydzielanych adipokin, jak również wykazywały odmienne wartości korelacji pomiędzy badanymi adipokinami a wskaźnikami gospodarki lipidowej w porównaniu do kobiet z nadwagą i otyłością z BMI 25-39,9 kg/m2.
2. U kobiet z otyłością olbrzymią zanotowano najwyższe wartości leptyny, wisfatyny i rezystyny wśród analizowanych grup kobiet z nadwagą i otyłością oraz w stosunku do grupy kontrolnej.
3. Kobiety z otyłością olbrzymią miały najniższe wartości adiponektyny całkowitej i wysokocząsteczkowej oraz aktywnej greliny w surowicy wśród analizowanych grup kobiet z nadwagą i otyłością, jak również w stosunku do grupy kontrolnej.
4. U pacjentek z otyłością olbrzymią wykazano odmienne niż w innych badanych grupach z nadwagą i otyłością korelacje pomiędzy leptyną a wskaźnikami gospodarki lipidowej (dodatnia korelacja leptyny z HDL-cholesterolem i ujemna z triglicerydami).
5. U kobiet z otyłością olbrzymią zanotowano też najsilniej wyrażony korzystny wpłwy wisfatyny na metabolizm cholesterolu (dodatnia korelacja wisfatyny z HDL-cholestreolem i ujemna z triglicerydami).
6. Po podzieleniu pacjentek z otyłością olbrzymią na grupę z obecnym zespołem metabolicznym i bez zespołu metabolicznego (metabolicznie zdrowych) stwierdzono znamiennie statystycznie wyższe wartości adiponektyny całkowitej i wisfatyny w grupie kobiet metabolicznie zdrowych. W grupie tej zanotowano także wyższe wartości HMW adiponektyny oraz niższe rezystyny w stosunku do kobiet z BMI ≥ 40 kg/m2 z towarzyszącym zespołem metabolicznym.
7. Porównanie grupy kobiet z BMI 25-39,9 kg/m2 z zespołem metabolicznym w stosunku do kobiet z BMI 25-39,9 kg/m2 bez zespołu metabolicznego wykazało znamiennie niższe wartości adiponektyny całkowitej i indeksu wolnej leptyny w grupie kobiet z BMI 25-39,9 kg/m2 skojarzonym z zespołem metabolicznym.
8. Stężenia adiponektyny całkowitej i HMW adiponektyny u kobiet z otyłością olbrzymią lub otyłością z BMI 25-39,9 kg/m2 bez towarzyszącego zespołu metabolicznego (metabolicznie zdrowych) były niższe niż u kobiet szczupłych z grupy kontrolnej, jednakże różnice te nie były istotne statystycznie.
9. Wykazano obecność istotnych korelacji pomiędzy stężeniami badanych adipokin a wartościami parametrów antropometrycznych i wskaźnikami metabolicznymi oraz insulinooporności we wszystkich badanych grupach kobiet z nadwagą i otyłością.
10. Obecność zespołu metabolicznego zarówno w przypadku otyłości z BMI 25-39,9 kg/m2, jak i otyłości olbrzymiej wiązała się z wyższymi wartościami ciśnienia tętniczego i gorszymi wskaźnikami gospodarki węglowodanowej.
11. Kobiety z nadwagą i otyłością, w tym otyłością olbrzymią, którym towarzyszył zespół metaboliczny miały najwyższe wartości wskaźnika HOMA-IR oraz najsilniej wyrażone zaburzenia gospodarki lipidowej.
12. Przeprowadzone analizy wyników badań genetycznych pomiędzy pacjentkami z nadwagą i otyłością z lub bez zespołu metabolicznego nie wykazały istnienia istotnych statystycznie różnic pomiedzy powyższymi grupami.
13. W przypadku polimorfizmu 276 G/T genu adiponektyny stwierdzono, że obecność allelu T w genotypie pacjentek z otyłością olbrzymią lub nadwagą i otyłością (BMI 25-39,9 kg/m2) skojarzoną z zespołem metabolicznym może wiązać się z 2,5-krotnie mniejszym ryzykiem wystapienia otyłości olbrzymiej lub nadwagi i otyłości skojarzonej z zespołem metabolicznym w porównaniu do młodych szczupłych kobiet.
14. W zakresie polimorfizmu rezystyny 420 C/G obecność allelu G u pacjentek z nadwagą i otyłością z zespołem metabolicznym w stosunku do grupy szczupłych kobiet z prawidłowym BMI wiąże się z ponad 2-krotnym wzrostem ryzyka wystąpienia zespołu metabolicznego.
15. Wykazano możliwość asocjacji polimorfizmów adiponektyny 276 G/T, 11 377 C/G i rezystyny 420 C/G z zespołem metabolicznym analizując grupy pacjentek z nadwagą i otyłością z zespołem metabolicznym w stosunku do kobiet szczupłych.
Przedstawione wyniki badań kobiet z nadwagą i otyłością miały charakter kompleksowy ponieważ dokonano oceny wydzielania licznych adipokin i peptydów, związanych z kontrolą łaknienia w odniesieniu do parametrów antropometrycznych i wyników badań biochemicznych. Badano także potencjalne asocjacje polimorfizmów genów adiponektyny i rezystyny w odniesieniu do otyłości i zespołu metabolicznego.
W wyniku przeprowadzonych badań stwierdzono znaczne różnice w profilu wydzielanych adipokin pomiędzy pacjentkami z nadwagą i otyłością skojarzoną z zespołem metabolicznym w odniesieniu do pacjentek z BMI ≥ 25 kg/m 2 bez zespołu metabolicznego (metabolicznie zdrowych). Wyodrębniono także adipokiny, które mogą być potencjalnym markerem zaburzeń metabolicznych w otyłości, co może mieć w przyszłości zastosowanie kliniczne.
Wnioski
1. Kobiety z otyłością bez zespołu metabolicznego (tzw. metabolicznie zdrowe) wykazywały istotne różnice w wydzielaniu adipokin w porównaniu do kobiet z otyłością skojarzoną z zespołem metabolicznym.
2. Pacjentki z otyłością olbrzymią skojarzoną z zespołem metabolicznym wykazywały najbardziej nasilone zaburzenia w wydzielaniu badanych adipokin.
3. Spośród badanych adipokin adiponektyna i jej frakcja wysokocząsteczkowa (HMW), wydają się być najlepszymi markerami zaburzeń metabolicznych w otyłości. Wyższe stężenia adiponektyny i jej frakcji HMW mogą być czynnikami zapobiegającymi wystąpieniu zespołu metabolicznego.
4. Współistnienie zespołu metabolicznego i towarzysząca mu insulinooporność, wydaje się mieć większy wpływ na wydzielanie adipokin niż stopień klinicznego zaawansowania otyłości mierzony przy pomocy BMI oraz BIA.
5. Analizowane polimorfizmy genów adiponektyny i rezystyny nie wykazały asocjacji z nadwagą i otyłością skojarzoną z zespołem metabolicznym w odniesieniu do nadwagi i otyłości bez zespołu metabolicznego.
6. Badania peptydowe u osób z otyłością olbrzymią mogą być pomocne w podejmowaniu decyzji, dotyczących kompleksowego leczenia tych osób, a w szczególności rozważenia operacji bariatrycznej.
Introduction
Background
Obesity is a chronic metabolic disease resulted from dysfunction of energy homeostasis mechanisms. It is caused by disturbed appetite control that leads to increased fat mass, and in consequence to development of multi-organ disorders (including cardiovascular disease, bone and joint disorders, hyperlipidemia etc.).
Both overweight and obesity create an essential problem from the sociological and epidemiological points of view as there is an increasing number of cases particularly in well-developed countries.
Metabolic syndrome
Long-time follow-up of obese patients resulted in emerging of metabolic syndrome that was defined in the 80s of the 20th century. In details, hyperinsulinism correlated with insulin resistance is playing a fundamental role in pathophysiology of metabolic syndrome. This syndrome is associated with visceral obesity, dyslipidemia, hypertension and malfunction of glucose metabolism.
In case of metabolic syndrome there is an increased risk of developing type 2 diabetes (by 3 times) as well as cardiovascular disease.
Adipose tissue as an endocrine organ
Since 1994, when leptin and its receptors were discovered by Friedman et al., adipose tissue no more has been considered as energy reservoir only. Extensive research on adipose tissue revealed the existence of many substances secreted by adipocytes that are called adipokines (or adipose derived hormones). Moreover, the metabolic activity of adipose tissue has also been confirmed. Adiponectin, its high molecular weight fraction, (HMW adiponectin), resistin, wisfatin and leptin are the most important adipokins.
Adiponectin is a 33kDa polypeptide, synthesized in adipose tissues. Adiponectin concentration is increased due to body mass reduction. On the other hand, BMI rise is correlated with a decrease in serum adiponectin level.
Worth noticing is an influence of adiponectin on reduction of insulin resistance. Adiponectin is not only antidiabetic factor, but also has anti-inflammatory and anti-atherogenic properties.
Clinical trials showed lower adiponectin concentration in patients with coronary heart disease.
Recently, it has been discovered that adiponectin may exist in different serum forms: dimer, trimer, hexamer or multimer. High molecular weight (HMW) adiponectin seems to be the most biological active form of adiponectin and has aprox.16 subunits. Elevation in serum HMW adiponectin concentration or HMW to total adiponectin ratio correlate positively with enhanced insulin sensivity.
However, the exact role of adiponectin in physiology and pathophysiology has not been clarified yet. Some studies suggested that elevated total adiponectin levels are negative predictors of mortality in patients with cardiovascular disease or heart failure. On the other hand, adiponectin has positive impact on metabolism. Thus, assessment of adiponectin role in pathological conditions needs further studies.
Resistin is a protein secreted mainly by adipocytes. Moreover, its expression was also shown in blood immune cells. Enhanced resistin levels were found in the animal model of obesity. It could be suggested that high level of insulin resistance due to resistin action is caused by stimulation of gluconeogenesis and glycogenolysis. Resistin may also influence a deterioration of glucose utilization in skeletal muscles.
Visfatin is a polypeptide secreted by visceral fat. It has been suggested that it exerts insulin-mimetic effects. Elevated visfatin levels were found in cases of obesity and insulin resistance. Moreover, increased concentration of visfatin was reported in patients with chronic kidney diseases as visfatin may enhance malfunction of renal endothelium.
However, the role and mechanism of visfatin action are not known in details.
Leptin is a 16 kDa peptide hormone. It is considered as the most important adipokine. The major source of leptin is white adipose tissue, but it also could be produced but in smaller amounts by brown adipose tissue. It plays a significant role in a control of energy homeostasis by suppressing central appetite regulation centers in the hypothalamus. Post-feeding elevation of circulating leptin results in decrease of neuropeptide Y (NPY) levels. Generally obese subjects have high levels of leptin that could be due to leptin resistance.
Studies published recently concerning the relation between leptin and atherosclerosis have indicated that leptin enhances endothelial dysfunction, activates oxidative stress and inflammatory processes, increases platelets aggregation and modulates smooth muscles proliferation.
Role of neuropeptides, secreted in the central nervous system and peripherally, in appetite control
Malfunction of appetite regulatory processes results in development of overweight and obesity. Eating behavior is regulated by neuropeptides secreted in the central nervous system and in gastrointestinal track, as well as by adipocyte derived hormones. The main regulatory organ for food intake is hypothalamus, where the hunger and satiety centers are found.
Neuropeptides involved in the regulation of food intake are divided into two opposing groups: orexigenic peptides and anorexigenic peptides. The group of orexigenic peptides consists of peptides derived from the central nervous system (neuropeptide Y, orexins, Agouti related peptide, galanin, endogenous opioids and endocannabinoids), and produced peripherally (the major role plays ghrelin – a peptide secreted in the gastrointestinal track, mainly by the stomach cells). Among anorexigenic peptides there are centrally produced: cocaine-amphetamine regulated transcript (CART), proopiomelanocortin (POMC), corticotrophin releasing factor (CRF) and serotonin, and released from the gastrointestinal track: cholecystokinin, peptide YY (PYY), amylin, insulin and bombesin, and in addition leptin secreted by the adipose tissue.
Peptides produced peripherally
Besides the central regulation, the peripheral activity is also important in the control of food intake. The most important gastrointestinal peptides are ghrelin possessing orexigenic properties and cholecystokinin that is an anorexigenic peptide.
Briefly, ghrelin is 28 aminoacids peptide that was firstly isolated from rat stomach. To date other organs have been reported to secrete ghrelin and that includes pituitary, hypothalamus, pancreas, lungs and immunological system. It has been reported that ghrelin levels are elevated during fasting and high concentration of ghrelin is a stimulus of food intake.
Hormonal disturbances in obesity
Long-lasting observations have confirmed that hormonal system is involved in appetite control and energy homeostasis. One of the most important hormones is insulin having extensive effect on metabolism and food intake regulation. Insulin increases: glucose oxidation, fatty acid synthesis, aminoacids uptake; and it decreases: lipolysis, lipid oxidation and gluconeogenesis.
The characteristic feature of a large population of obese people is insulin resistance, connected with hiperinsulinism. Reduction of body mass being a result of diet and/or pharmacotherapy or bariatric surgery leads to decrease in the fat mass as well as to reduce insulin resistance.
In addition, disturbances in the thyroid and adrenal function may also contribute to obesity.
Genetic factors and obesity
It is known that obesity is correlated with many environmental and genetic factors. Studies investigating families of obese subjects revealed that genetic factors impact on BMI in 30-40% and environmental influence is estimated at approx. 60-70%.
Undoubtedly, the genetic factors condition predisposition to obesity. However, generally obesity should not be considered as a genetic disorder only. It is known that genetic determinants of obesity may be divided into single gene or polygenic disorder. The best recognized gene mutations among single gene disorders are mutations of melanocortin 4 receptor gene, leptin gene and leptin receptor gene. Nevertheless, it is speculated that polygenic heredity is a significant cause of obesity.
Recently, it has been published that polymorphism of FTO (Fat mass and obesity-associated gene) may be another factor of great importance contributing to obesity. European studies on children and youngsters indicate that some polymorphisms of FTO are associated with the weight gain that is due to larger amounts of consumed food and deprivation of postprandial satiety. Besides, data concerning the role of FTO are ambiguous and suggest the necessity of further studies.
Another genetic factor that may contribute to development of obesity is PPAR γ (peroxisome proliferator-activated receptor γ) gene. Investigations conducted on PPAR γ gene polymorphism resulted in conclusion that polymorphism pro12ala may be associated with lower risk of type 2 diabetes, enhanced insulin sensitivity and decreased BMI.
Genetic disturbances resulting in impropriate synthesis and malfunction of adipokin may also contribute to obesity. Polymorphisms of adiponectin gene considered to be important are as follows: single nucleotide polymorphism (SNP) in the promotor region, SNP 45 T/G in the signal sequence and polymorphism of introns. It is suggested that polymorphisms mentioned above may increase the risk of type 2 diabetes, insulin resistance and hypertension. Thus, they may lead to metabolic syndrome.
Resistin is an adipokine, probably associated with insulin resistance in a course of obesity and metabolic syndrome. Increased serum/plasma resistin concentration has been found in obese patients suffering from diabetes. The most often described polymorphism of resistin is 420 C/G (located in the promotor region). It is associated with the presence of obesity, insulin resistance and type 2 diabetes in different populations. However, the results of studies on the exact role of resistin gene polymorphism in development of metabolic syndrome are still equivocal and further investigation is needed.
The goal of the study
Despite the fact that to date data from many studies conducted on metabolic syndrome and obesity have been published, there are still questions concerning pathophysiology and medical consequences of those medical conditions. Along with the growth of clinical stages of obesity, measured with BMI, subsequently the number of subjects with metabolic syndrome is increasing. However, metabolic syndrome is not expressed in all obese patients, even those with morbid obesity (with BMI ≥ 40 kg/m2). This particular group of patients is named metabolically healthy obese. According to the findings of severe obesity not coexisting with metabolic disturbances, type 2 diabetes or hypertension, it should be taken into a consideration if there are any differences in the adipokine profile (possibly with genetic associations) between obese individuals with and without metabolic syndrome. Unambiguous changes in the adipokine profile or variants of genetic predictors involved in development of metabolic syndrome have not yet been established.
In the present study, it has been attempted to assess genetic factors using analyses of polymorphism frequency of two adipokines with opposing action: adiponectin (that decrease insulin resistance and has anti-atheroslerotic and antidiabetic properties) and resistin (that is thought to stimulate insulin resistance). Finding of statistically significant differences between occurrence of studied polymorphism genotypes or their associations with metabolic syndrome could be probable predictor of metabolic disturbances in obesity.
In addition, a combination of research concerning changes in the adipokine profile corresponding with the insulin resistance assessment and studies on polymorphisms of adiponectin and resistin genes may help to emerge the group of patients with particularly enhanced metabolic disturbances who need an intensive medical supervision.
The aim
The aim of the study is to distinguish factors influencing the development of metabolic syndrome in overweight or obese individuals.
Changes in adipokine profile as well in concentration of peptides involved in the control of appetite will be analyzed in groups of patient with different stages of obesity, with or without metabolic syndrome.
Moreover, additional research will be performed to assess correlations between peptide levels and insulin resistance index, lipid profile and anthropometric parameters.
Besides, association of gene polymorphism (adiponectin and resistin genes) with metabolic status of overweight or obese individuals will also be studied.
Results from this study will allow to select peptide or genetic markers of development of metabolic syndrome.
Research tasks
1) Assessment of anthropometric parameters, lipid profile and carbohydrate metabolism in following groups of women:
a) Lean, healthy women with BMI <25 kg/m2,
b) Overweight or obese women (BMI 25-39,9 kg/m2) with metabolic syndrome,
c) Overweight or obese women (BMI 25-39,9 kg/m2) without metabolic syndrome,
d) Women with morbid obesity (BMI> 40 kg/m2).
2) Analyses of changes in the adipokine profile and peptide concentrations (leptin, total adiponectin, HMW adiponectin, visfatin, ghrelin, resistin) in all groups in comparison with control group and between particular groups.
3) Evaluation of differences in the adipokine profile, peptide concentrations and biochemical parameters in individuals with metabolic syndrome compared to those without.
4) Assessment of correlations and relationship between changes in adipokine concentrations and other parameters (anthropometric parameters, lipid profile, carbohydrate metabolism parameters) in examined groups of overweight/obese women.
5) Evaluation of gene polymorphism frequency (for adiponectin and resistin genes) and establishing the Odds Ratio (OR) in following groups:
a) Overweight/obese women with metabolic syndrome compared to overweight/obese women without metabolic syndrome,
b) Overweight/obese women with metabolic syndrome compared to lean, healthy women from control group,
c) Overweight/obese women without metabolic syndrome compared to lean, healthy women from control group.
6) Determination of associations between metabolic syndrome and analyzed polymorphisms.
7) Finding of peptide and/or genetic markers suitable for diversifying overweight/obesity with metabolic syndrome and overweight/obesity without metabolic syndrome.
Material and methods
The study was carried out on 369 women who were assigned to following groups:
? 34 women with morbid obesity (BMI> 40 kg/m2),
? 100 overweight/obese women (BMI 25-39,9 kg/m2) with metabolic syndrome,
? 131 overweight/obese women (BMI 25-39,9 kg/m2) without metabolic syndrome,
? 04 young, healthy, lean women (BMI 18-24,9 kg/m2) as a control group.
Metabolic syndrome was diagnosed according to criteria of International Diabetic Federation (2005). Hypertension was defined in accordance with ESH and ESC criteria. Diagnosis of type 2 diabetes was established when fasting plasma glucose was ≥ 126 mg/dl or plasma glucose concentrations were ≥ 200 mg/dl two hours after 75 g oral glucose load (oral glucose tolerance test) or when anytime plasma glucose was ≥ 200 mg/dl. Classification of diabetes was assessed in accordance with criteria of World Health Organization Experts Committee on Diabetes (1998).
Excluding criteria were as follows:
? Endocrine diseases
? Chronic pulmonary dysfunction
? Chronic kidney and liver diseases
? Neoplasm history
None of the examined subject had signs of acute infectious disease at the time of the investigation. None of them was taking anti-inflammatory drugs. History of excessive alcohol consumption and/or smoking eliminated individuals from the study.
The study protocol was accepted by Bioethical Committee of Medical Centre of Postgraduate Education.
Informed consent was obtained from all the subjects.
Medical examination
Clinical data including blood pressure, height, weight and BMI calculation were collected. Waist measurement and waist-hip ratio (WHR) were applied as an indicator of central obesity. The fat content was measured with electric bioimpedance (BIA).
Peptide and hormone measurements. Assessment of lipid and glucose profile
Blood samples were taken from all subjects in the morning hours after overnight (12 hrs) fasting. Immediately after collection, the samples were centrifugated at temperature of 4°C, and then obtained serum was frozen at -40°C.
Leptin, adiponectin and ghrelin active concentrations were estimated using RIA methods.
Resistin and soluble leptin receptor levels were measured by ELISA tests.
C-end visfatin fragments and high molecular weight (HMW) adiponectin levels were established with EIA methods.
Insulin concentration was measured using IRMA tests.
Measurements of lipid and glucose profile were made with routine laboratory tests.
Insulin resistance was estimated using the homeostasis model assessment method (HOMA-IR).
Intra- and inter assay coefficient was under 10% for all investigated parameters.
Genotyping of selected polymorphisms of adiponectin and resistin genes
Genomic DNA was extracted from whole blood treated with EDTA using NucleoSpin Blood Quick Pure kit (Macharey-Nagel, Germany) in accordance with the kit´s protocol.
The following single nucleotide polymorphism (SNP) for resistin gene were investigated: 420 C/G, 62 G/A, 537 A/C, and respectively for adiponectin gene: 45 T/G, 276 G/T, 11 377 C/G, 11 391 G/A.
Genotyping was carried out using TaqMan SNP Genotyping Assays. The Real Time PCR (RT-PCR) was performed with use of ABI PRISM Sequence Detection System (SDS). Allelic discrimination was carried out using ABI PRISM 7000SDS Software.
Statistical analyses
Statistical analyses were performed using STATISTICA ver. 7.1 PL software. The statistical significance was accepted at 0.05.
The presence of normality in distribution among the groups was investigated by the Shapiro-Wilk test and the Kolmogorow-Smirnov test with the Lilliefors correction.
Evaluation of differences between groups was performed using the Kruskal- Wallis rank test, followed by the Mann-Whitney U-test. To calculate correlation coefficients between adipokines and data from anthropometric examination and biochemical parameters, the Spearman test was applied.
Analysis of covariance (ANCOVA) was performed in obesity and overweight groups. Adiponectin, HMW adiponectin, leptin, resistin or visfatin, respectively were adjusted for BMI, BIA and WHR.
Results of peptide and adipokine concentrations, anthropometric parameters and lipid profile were expressed as mean ± SD.
The comparison of frequency distribution of polymorphism genotype of adiponectin and resistin genes was performed using the χ2 test. OR with 95% confidence intervals (95% CI) were calculated. Evaluation was carried out for dominant model (heterozygote + recessive homozygote) vs. dominant homozygote.
Calculation of the Hardy-Weinberg equilibrium was also estimated.
To establish associations between analyzed adiponectin and resisting gene polymorphisms with overweight, obesity and metabolic syndrome logistic regression was used. In addition, simultaneously OR was evaluated. The logistic regression was performed between following groups:
1. Overweight/obese women with metabolic syndrome compared to overweight/obese women without metabolic syndrome.
2. Overweight/obese women with metabolic syndrome compared to young, lean women (BMI within the normal range).
3. Overweight/obese women without metabolic syndrome compared to young, lean women (BMI within the normal range).
Results
Outcomes from the investigation are as follows:
1. Subjects suffering from morbid obesity had the most visible disturbances in adipokine release as well as showed different pattern of correlation between adipokines and lipid parameters when compared with obese women with BMI 25-39,9 kg/m2.
2. Women with morbid obesity had the highest concentrations of leptin, visfatin and resistin in comparison with groups of overweight/obese subjects as well as with the control group.
3. Women with morbid obesity had the lowest levels of total adiponectin, HMW adiponectin and ghrelin among all investigated groups and when compared with the controls.
4. Morbid obese patients, when compared with overweight/obese group, showed different correlations between leptin and lipid parameters (positive correlation between leptin and HDL cholesterol and negative correlation between leptin and triglycerides).
5. Different pattern of correlations between visfatin and lipid parameters were found in morbid obese group when compared with all other examined groups. In details, a positive correlation between visfatin and HDL cholesterol, and negative one between visfatin and triglycerides was found.
6. Analysis of individuals with morbid obesity, divided into two subgroups of metabolic healthy and those with metabolic syndrome, revealed that in group of metabolic healthy subjects statistically significant higher values of total adiponectin and visfatin were found. In addition, higher concentrations of HMW adiponectin and lower levels of resistin were reported in morbid obese but metabolically healthy women.
7. Comparison of patients with BMI 25-39,9 kg/m2 with or without metabolic syndrome showed that statistically significant lower values of total adiponectin, leptin and lower free leptin index were found in women with metabolic syndrome.
8. In obese women (including those with morbid obesity) suffering from metabolic syndrome values of total and HMW adiponectin had tendency to be higher when compared to lean women. However, these differences were not statistically significant.
9. Significant correlations were found between adipokines and anthropometric features, metabolic parameters and insulin resistance markers.
10. The coexistence of metabolic syndrome and obesity (including morbid obesity) resulted in findings of higher blood pressure measurements and worse carbohydrate metabolism parameters.
11. Women being overweight or obese (including those with morbid obesity) with diagnosed metabolic syndrome had the highest values of HOMA-IR and presented the most intensive disturbances in lipid profile.
12. Investigated allelic distribution in groups of overweight/obese women with or without metabolic syndrome did not show statistically significant differences between those groups.
13. The presence of allele T in 276 G/T polymorphism of adiponectin gene in patients with overweight or obesity, in whom metabolic syndrome was diagnosed, may be associated with 2.5 lower risk of development of morbid obesity or obesity with metabolic syndrome when compared to lean young women.
14. The presence of allele G in 420 C/G polymorphism of resistin gene in groups of overweight/obese women diagnosed with metabolic syndrome may suggest that there is more than 2 times higher risk of metabolic syndrome development in comparison with lean young women.
15. The possible associations of 276 G/T, 11 377 C/G adiponectin gene polymorphism and 420 C/G resistin gene polymorphism with metabolic syndrome were found when comparing overweight/obese subjects being diagnosed with metabolic syndrome to lean women.
Data showed above concerning women with overweight/obesity give a complex characteristic of adipokine and peptide (important in the appetite control) release that was referenced to anthropometric features and biochemical parameters. Moreover, the evaluation of potential associations of adiponectin and resistin gene polymorphisms in case of obesity and metabolic syndrome was also conducted.
The study resulted in findings of the significant differences in adipokine release pattern when compared individuals with overweight/obesity suffering from metabolic syndrome with adequate subjects without metabolic disturbances (metabolic healthy).
It should be highlighted that some adipokines, selected in this study, may be markers of metabolic abnormalities and in consequence, estimation of those adipokines may have therapeutic implication in future.
Conclusions
1. Obese women without metabolic syndrome (so called metabolically healthy) presented a significantly different adipokine pattern when compared to obese subjects without metabolic syndrome.
2. Subjects with morbid obesity that coexisted with metabolic syndrome had the most disturbed adipokine release.
3. Among analysed adipokines, adiponectin and its HMW form seem to be the best markers of metabolic disturbances in obesity. Incresed values of total and HMW adiponectin may act as factors preventing from development of metabolic syndrome.
4. The coexistence of metabolic syndrome, associated with insulin resistance, may have stronger impact on adipokine release than clinical stage of obesity measured by BMI and BIA.
5. Investigated polymorphisms of adiponectin and resistin genes did not show associations with overweight and obesity combined with metabolic syndrome when compared to overweight and obesity without metabolic syndrome.
6. The evaluation of peptide concentrations in patients with morbid obesity may be helpful in decision making processes concerning the treatment methods, especially when bariatric surgery is considered.
WSTĘP
Otyłość – definicja i dane epidemiologiczne
Otyłość jest przewlekłą chorobą metaboliczną wynikającą z zaburzeń homeostazy energetycznej organizmu (1). Spowodowana jest zaburzeniem kontroli łaknienia, co prowadzi do zwiększenia ilości tkanki tłuszczowej, a w konsekwencji do rozwoju powikłań wielonarządowych (w tym chorób układu sercowo-naczyniowego, kostno-stawowego, zaburzeń gospodarki lipidowej). Graniczną wartością, od której rozpoznawana jest otyłość, jest przekroczenie tzw. wskaźnika masy ciała ponad 30 kg/m2 (wskaźnik masy ciała; ang. body mass index), co stanowi około 120% prawidłowej (tj. należnej) masy ciała. Wartości 110-120% należnej masy ciała odpowiadają BMI odpowiednio 25-30 kg/m2 i określane są jako nadwaga (1, 2, 3).
Nadwaga i otyłość stanowią istotny problem epidemiologiczny ze względu na dynamiczny wzrost częstości ich występowania, przede wszystkim w krajach wysokorozwiniętych. Szacuje się obecnie, że ok. 30% mieszkańców Stanów Zjednoczonych ma BMI ≥ 30 kg/m2 i wartości te wykazują stałą tendencję wzrostową. Nadwaga w populacji amerykańskiej w 2000 roku stwierdzana była u 56,4% osób dorosłych (65,5% mężczyzn i 47,6% kobiet). Dane te są alarmujące nie tylko z powodu wzrostu ryzyka powikłań związanych z otyłością, ale przede wszystkim ze względu na dynamikę rozprzestrzeniania się choroby, albowiem w 1991 roku ilość osób z nadwagą szacowana była w USA na ok. 45% (1, 2, 4, 5, 6).
Kolejnym istotnym problemem epidemiologicznym jest fakt stałego zwiększania się częstości występowania nadwagi i otyłości u dzieci i młodzieży (7). W ciągu ostatnich 2-3 dekad wykazano wzrost częstości nadwagi i otyłości w Brazylii z 4,1% do 13,9%, w Chinach z 6,4% do 7,7%, a w Stanach Zjednoczonych z 15,4% do 25,6%. Jedynym krajem, w którym stwierdzono spadek częstości występowania nadwagi i otyłości u dzieci i młodzieży, była Rosja (8).
W populacji polskiej częstość BMI ≥ 30 kg/m2 waha się pomiędzy 12 a 20% a BMI 25-29 kg/m2 i wynosi 30-40%. Wśród dzieci i młodzieży do lat 18 wartości te wynoszą odpowiednio 5-8% i 8-12% (1, 7), jednakże badania epidemiologiczne prowadzone w różnych regionach w Polsce wskazują na duże różnice lokalne. Najczęściej nadwaga i otyłość u dzieci i młodzieży występuje w miastach w województwach zachodniej i centralnej Polski (7, 9, 10, 11).
Przedstawione dane epidemiologiczne wskazują na znacznie zwiększone ryzyko wystąpienia chorób układu krążenia, oddechowego, zespołu metabolicznego, cukrzycy typu 2, nowotworów czy chorób zwyrodnieniowych układu kostnego. Istotnym problemem klinicznym u osób otyłych jest pogorszenie jakości życia, co wiąże się, między innymi z problemami w uzyskaniu zatrudnienia lub możliwości awansu zawodowego.
Wymienione poprzednio przewlekłe schorzenia skojarzone z otyłością prowadzą do wzrostu umieralności (w Stanach Zjednoczonych umiera każdego roku ok. 300 000 osób z powodu chorób przewlekłych spowodowanych otyłością (12)) i skrócenia tzw. średniego oczekiwanego okresu życia. Ten ostatni fakt jest szczególnie ważny w przypadku dzieci i młodzieży. Must i wsp. (13) wykazali, że otyłość u dzieci i młodzieży koreluje ze wzrostem zachorowalności i śmiertelności po 50. latach życia niezależnie od masy ciała tych osób w wieku dorosłym. Szacuje się, że otyłość brzuszna zmniejsza średni oczekiwany okres życia u mężczyzn o ok. 20-25%, a u kobiet o 15-20% (1).
Nadwaga i otyłość stanowią obecnie istotny problem społeczny pogarszający nie tylko jakość życia osób chorych, ale są również ważnym problemem ekonomicznym w wielu krajach świata, w tym także w Polsce.
Zespół metaboliczny
Wieloletnie obserwacje kliniczne osób otyłych doprowadziły do wyodrębnienia i zdefiniowania pod koniec lat osiemdziesiątych XX-tego wieku zespołu metabolicznego, którego patogenetycznym podłożem jest hiperinsulinizm z insulinoopornością. Zespół ten skojarzony jest z otyłością trzewną, dyslipidemią, nadciśnieniem tętniczym i zaburzeniami gospodarki węglowodanowej (14, 15, 16). Obecnie przyjęte kryteria rozpoznania tego zespołu opierają się na ustaleniach IDF (International Diabetes Federation) z 2005 roku i NCEP-ATP III (National Cholesterol Education Program – Adult Treatment Panel III) z 2001 roku. Częstość występowania tej jednostki chorobowej rośnie wraz z wiekiem i oceniana jest wśród amerykańskich mężczyzn w wieku 60-69 lat na 45%, a wśród kobiet na 42%. Dane epidemiologiczne uzyskane w badaniach innych populacji są mniej pesymistyczne i wahają się między 10 a 30% (1,14,15,16). Częstość występowania zespołu metabolicznego wzrasta także ze wzrostem BMI, jednakże nawet u osób z otyłością olbrzymią (BMI ≥ 40 kg/m2) nie występuje on u wszystkich chorych.
Najistotniejszym problemem klinicznym w skali populacyjnej jest fakt, że zespół metaboliczny zwiększa ponad 3-krotnie ryzyko cukrzycy typu 2 oraz chorób układu sercowo-naczyniowego (15, 16, 17, 18).
Przedstawione powyżej dane epidemiologiczne wskazują na bardzo duże znaczenie społeczne otyłości i jej konsekwencji zdrowotnych.
Mechanizmy patofizjologiczne dotyczące otyłości nie są wciąż jednoznacznie wyjaśnione. Należy jednak pamiętać, że przez ostatnie kilkanaście lat wiedza na temat przyczyn i podłoża otyłości została znacznie poszerzona. Jednym z takich faktów było odkrycie w 1994 roku leptyny, co w konsekwencji spowodowało uznanie tkanki tłuszczowej za narząd wydzielania wewnętrznego.
Tkanka tłuszczowa – narząd wydzielania wewnętrznego
Od momentu odkrycia w 1994 roku przez Friedmana i wsp. leptyny oraz jej receptorów (19, 20), tkanka tłuszczowa przestała być uważana jedynie za rezerwuar energetyczny ustroju, lecz stała się obiektem wnikliwych badań, które w konsekwencji doprowadziły do odkrycia licznych adipokin (substancji wydzielanych przez adipocyty), jak również wykazały istotną aktywność metaboliczną komórek tkanki tłuszczowej. Budowa tkanki tłuszczowej jest zróżnicowana, gdyż składa się ona nie tylko z samych adipocytów, ale także w jej skład wchodzą macierz (matrix), komórki autonomicznego układu nerwowego, komórki podścieliska i komórki immunologicznie kompetentne. Tkankę tłuszczową charakteryzuje ponadto bogate unaczynienie (21).
Procesy metaboliczne zachodzące w tkance tłuszczowej podlegają regulacji, m.in. przez hormony zarówno białkowe, jak i sterydowe oraz cytokiny, które wywierają działanie na szereg specyficznych receptorów zlokalizowanych na adipocytach. Do najważniejszych receptorów zalicza się:
1. Receptory błonowe dla insuliny, glukagonu, hormonu wzrostu, TSH oraz peptydu glukagonopodobnego typu 1 (GLP-1) i angiotensyny 1 i 2.
2. Receptory jądrowe dla glikokortykoidów, witaminy D3, hormonów tarczycy, androgenów, estrogenów i progesteronu.
3. Receptory dla cytokin i leptyny.
4. Receptory dla katecholamin (β1, β2, β3, α1, α2) (22, 23).
Obecnie wyróżnia się dwa główne rodzaje tkanki tłuszczowej, jednakże podział ten w świetle obecnych danych wydaje się być niedoskonały i w przyszłości będzie ulegał modyfikacji (21, 24):
1. Biała tkanka tłuszczowa, w której dominują adipocyty o średnicy 100-200 ?m zawierające w 95% triglicerydy. Tkanka ta pełni funkcję, przede wszystkim magazynu energetycznego ustroju pod postacią zdeponowanych triglicerydów. Biała tkanka tłuszczowa odpowiada za utrzymanie na odpowiednim poziomie stężenia wolnych kwasów tłuszczowych. Wydziela ona liczne adipokiny (między innymi adiponektynę, leptynę, rezystynę) biorące udział w procesach metabolicznych węglowodanów i lipidów. Wpływa pośrednio na kontrolę ciśnienia tętniczego przez syntezę angiotensynogenu, moduluje procesy immunologiczne (adiponektyna, leptyna) oraz koagulologiczne (wydziela inhibitor tkankowego aktywatora plasminogenu) (21, 22, 23, 24).
2. Brunatna tkanka tłuszczowa, w której przeważają adipocyty o mniejszych wymiarach rozproszone w obrębie podścieliska. Tkanka ta jest silniej unerwiona przez układ współczulny oraz posiada system mitochondriów związany z białkiem rozsprzęgajacym UCP 1 (uncoupling protein 1). Brunatna tkanka tłuszczowa utylizuje lipidy z wytworzeniem energii. Pod wpływem pobudzenia układu współczulnego i receptorów PPARγ może dochodzić do przekształcania się tkanki tłuszczowej białej w brunatną (21, 22, 23, 24, 25).
Poza przedstawionym podziałem tkankę tłuszczową możemy również podzielić na tkankę podskórną i brzuszną (trzewną). Tkanka tłuszczowa trzewna w porównaniu do podskórnej syntetyzuje i wydziela do krwiobiegu więcej cytokin prozapalnych (TNFα, IL6), posiada więcej receptorów androgenowych, receptorów β adrenergicznych (szczególnie β3) i glikortykoidowych, jak również więcej wydziela rezystyny (22, 26).
Niekorzystne efekty metaboliczne wynikające ze zwiększenia objętości trzewnej tkanki tłuszczowej związane są nie tylko ze zwiększoną syntezą niektórych adipokin, wolnych kwasów tłuszczowych, ale również z ich bezpośrednim wnikaniem do krążenia wrotnego i pominięciem metabolizowania w wątrobie (brak tzw. efektu pierwszego przejścia). Należy podkreślić negatywną rolę wolnych kwasów tłuszczowych (WKT), które nasilają glukoneogenezę, zaburzają metabolizm insuliny, stymulują produkcję triglicerydów i apolipoproteiny B. Dodatkowo cytokiny prozapalne syntetyzowane w adipocytach stymulują wydzielanie białka C reaktywnego (21, 27).
Istotnym czynnikiem związanym z patofizjologicznym działaniem tkanki tłuszczowej trzewnej jest aktywność znajdującej się w niej 11β dehydrogenazy hydroksysteroidowej typu 1 przekształcającej nieaktywny kortyzon w kortyzol. U osób otyłych aktywność tego enzymu jest znacznie podwyższona, co powoduje zwiększenie lokalnego działania kortyzolu (21, 22, 28).
Wszystkie opisane powyżej mechanizmy patofizjologiczne prowadzą do rozwoju insulinooporności i w konsekwencji do wystąpienia zespołu metabolicznego (14, 15, 26, 29, 30).
Tkanka tłuszczowa jest źródłem wielu substancji biologicznie czynnych. W poniższej tabeli przedstawione zostały adipokiny, enzymy i inne czynniki syntetyzowane w tkance tłuszczowej (22, 23).
Tabela 1. Substancje biologicznie czynne wydzielane przez tkankę tłuszczową.
Adiponektyna
Adiponektyna jest polipeptydem o masie cząsteczkowej 33 kDa syntetyzowanym przez tkankę tłuszczową. Białko to jest kodowane przez gen zlokalizowany na 3 chromosomie (3q27), który składa się z 3 eksonów. Synteza i wydzielanie do krążenia obwodowego adiponektyny stymulowane są przez, między innymi agonistów receptora PPARγ, jak również jej stężenie w surowicy wzrasta wraz redukcją masy ciała (efekt ten prawdopodobnie spowodowany jest obniżeniem syntezy TNFα w tkance tłuszczowej, który działa hamująco na syntezę adiponektyny) (26, 31, 32). Natomiast wraz ze wrostem BMI stwierdzany jest spadek adiponektyny w surowicy krwi obwodowej. Adiponektyna występuje w postaci dimerów, trimerów, tetramerów oraz oligomerów.
Wywiera ona swoje działanie poprzez 2 typy receptorów (Adipo R1 i Adipo R2) związanych z białkiem G. Receptor typu 1 zlokalizowany jest w tkance tłuszczowej i mięśniach, a receptor typu 2, przede wszystkim w wątrobie (22, 26, 31, 33).
Niezwykle istotnym metabolicznie działaniem adiponektyny jest jej wpływ na zmniejszenie insulinooporności przez hamowanie glukoneogenezy wątrobowej, zwiększenie oksydacji kwasów tłuszczowych i zużycia glukozy oraz metabolizmu mleczanów w mięśniach. W wątrobie pod wpływem adiponektyny nasila się oksydacja kwasów tłuszczowych, co powoduje zmniejszenie stężenia wolnych kwasów tłuszczowych i triglicerydów w surowicy. Poza korzystnym działaniem antydiabetogennym, przeciwzapalnym (adiponektyna hamuje syntezę cytokin prozapalnych) peptyd ten może wykazywać działanie przeciwmiażdżycowe. Hamuje on adhezję monocytów do śródbłonka naczyniowego, zmniejsza ekspresję cząsteczki adhezyjnej VCAM1 oraz selektyny E. Adiponektyna stymuluje także syntezę tlenku azotu w komórkach śródbłonka w mechanizmie wzrostu mRNA dla endotelialnej syntazy tlenku azotu (eNOS). Zwiększenie syntezy NO przez endotelium skutkuje rozkurczem mięśniówki gładkiej naczyń.
Istotnym klinicznie faktem jest obniżenie stężenia adiponektyny u pacjentów z chorobą niedokrwienną serca (22, 26, 31, 33).
Adiponektyna w surowicy może występować w postaci kilku frakcji, w tym: dimerów, trimerów, heksamerów lub multimetrów. Frakcja wysokocząsteczkowa adiponektyny (tzw. HMW adiponectin), składająca się z ok. 16 cząsteczek tego peptydu, wydaje się być najbardziej aktywną formą tej adipokiny. Wzrost poziomu HMW adiponektyny lub stosunku HMW adiponektyny do adiponektyny całkowitej koreluje dodatnio ze wzrostem insulinowrażliwości w przebiegu leczenia lekami z grupy tiazolidinedionów u pacjentów z cukrzycą typu 2. Wydaje się, że zwiększanie się stężenia wysokocząsteczkowej adiponektyny w surowicy oraz podwyższony stosunek HMW adiponektyny do adiponektyny całkowitej koreluje silniej ze stopniem obniżenia insulinooporności niż zmiany stężeń adiponektyny całkowitej. Jest zatem prawdopodobne, że u osób z zespołem metabolicznym stężenie HMW adiponektyny może być pośrednim markerem stopnia nasilenia insulinooporności (34).
Powyższe dane potwierdzają antydiabetogenne, przeciwzapalne i przeciwmiażdżycowe działanie tej adipokiny. Jednakże rola i znaczenie adiponektyny w wielu procesach patologicznych nie są do końca jednoznacznie wyjaśnione (35). Badania opublikowane przez Deckert i wsp. w 2008 r. (36) wskazują, że wyższe poziomy całkowitej adiponektyny mogą być negatywnym czynnikiem prognostycznym u osób z chorobami układu sercowo-naczyniowego, jak również w przewlekłej niewydolności krążenia (37). Z drugiej strony, w innych pracach wykazano, że u części pacjentów z otyłością olbrzymią (BMI ≥ 40 kg/m2) stwierdza się stężenia adiponektyny porównywalne z osobami z prawidłową BMI (38). Dodatkowo, badania własne oraz prace badaczy japońskich i amerykańskich przeprowadzone u osób powyżej 100 roku życia wykazały, że stężenia adiponektyny w grupie osób stuletnich były wyższe w porównaniu z wartościami uzyskanymi u osób młodych oraz z grupy kontrolnej w wieku ok. 70 lat. Badania te wykazały również mniejszą insulinooporność u osób powyżej 100 roku życia (39, 40, 41).
Rezystyna
Rezystyna (resistin to insulin) jest białkową adipokiną o masie cząsteczkowej 12 kDa i składa się ze 108 aminokwasów. Syntetyzowana jest w adipocytach, ale jej ekspresję stwierdzono także w komórkach jednojądrowych krwi obwodowej. Synteza rezystyny kodowana jest przez gen zlokalizowany w 19 chromosomie (19p13.2) posiadającym 4 eksony (22, 23, 26, 42, 43).
Ekspresja mRNA dla rezystyny hamowana jest przez tiazolidinediony. U zwierząt doświadczalnych stwierdzono 15-krotnie większą ekspresję rezystyny w wisceralnej tkance tłuszczowej w stosunku do podskórnej tkanki tłuszczowej (44). Podwyższone wartości rezystyny wykazano w zwierzęcym modelu otyłości, przy czym podanie zwierzętom rezystyny egzogennej nasilało insulinooporność (22, 45). Badania in vivo, jak i in vitro wskazują na wzrost stężenia rezystyny w przebiegu procesów zapalnych i insulinooporności (46, 47).
Wyniki badań dotyczących rezystyny są rozbieżne, gdyż część z opublikowanych dotychczas prac przedstawia wyniki przeciwne w stosunku do opisanych powyżej. Interpretacja badań eksperymentalnych prowadzonych na modelach zwierzęcych oraz ich ekstrapolacja w stosunku do populacji ludzkiej jest trudna do oceny, gdyż rezystyna ludzka i zwierzęca (u gryzoni) wykazują homologię budowy jedynie w 64% (44, 45, 48).
Wydaje się, że nasilanie insulinooporności przez rezystynę spowodowane jest stymulacją glukoneogenezy i glikogenolizy. Rezystyna ma również wpływ na pogorszenie utylizacji glukozy w mięśniach szkieletowych poprzez zmniejszenie ekspresji transportera GLUT4 (23).
Wisfatyna
Wisfatyna jest polipeptydem produkowanym, przede wszystkim przez trzewną tkankę tłuszczową. Początkowo wisfatynę określano jako PBEF (pre-β cell colony-enhancing factor), gdyż po raz pierwszy wykazano jej obecność w limfocytach. Poza tym może być syntetyzowana w śródbłonku naczyniowym, w sutku oraz płucach (23, 49, 50). Sugeruje się, że może ona wywierać działanie insulinomimetyczne (23, 49)
W badaniach in vitro wykazano, że ekspresja wisfatyny wzrasta pod wpływem deksametazonu. Jednakże nie potwierdzono tego faktu, badając stężenia tego peptydu po wykonaniu krótkiego testu hamowania z deksametasonem (49, 50). Podwyższone wartości wisfatyny stwierdzane są w otyłości, jak również w insulinooporności (49, 50, 51). W innych badaniach wykazano dodatnią korelację pomiędzy poziomem wisfatyny a stężeniem HDL-cholesterolu (52, 53). Ponadto podwyższone poziomy wisfatyny wykazano u chorych z przewlekłymi chorobami nerek (co można tłumaczyć zaburzoną eliminacją tej adipokiny lub może być odzwierciedleniem przewlekłego procesu zapalnego). W tej grupie chorych wisfatyna może nasilać dysfunkcję śródbłonka naczyniowego (54). Interesujący jest fakt, że wyższe w stosunku do grupy kontrolnej stężenia wisfatyny stwierdzane były także u pacjentów z narkolepsją (55).
Rola, jak również mechanizm działania wisfatyny, nie został jeszcze w pełni wyjaśniony i wymaga przeprowadzenia dalszych szczegółowych badań zarówno eksperymentalnych, jak również klinicznych.
Leptyna
Leptyna została odkryta w 1994 r. i jest białkiem o masie cząsteczkowej ok. 16 kDa zbudowanym ze 167 aminokwasów, syntetyzowanym przede wszystkim w adipocytach białej tkanki tłuszczowej oraz w niewielkich ilościach w brunatnej tkance tłuszczowej. Poza tym leptyna syntetyzowana jest także w łożysku, komórkach OUN oraz w komórkach gruczołowych żołądka (56).
Gen kodujący leptynę zlokalizowany jest u człowieka w chromosomie 7 (7q31), składa się z trzech eksonów podzielonych 2 intronami i zawiera 15 000-20 000 par zasad.
Leptyna wywiera swoje działanie przez specyficzne receptory homologiczne do glikoproteiny 130 (gp 130) zaliczanej do I klasy receptorów błonowych dla cytokin. Do chwili obecnej wyizolowano 5 izoform tego receptora, wśród których najważniejszymi są:
? Receptory posiadające długą domenę cytoplazmatyczną (ObRb). Receptory tego typu zlokalizowane są, przede wszystkim w podwzgórzu (szczególnie w jądrze łukowatym). Poprzez ten receptor przekazywane są informacje dla neuronów podwzgórzowych.
? Receptory posiadające krótką domenę cytoplazmatyczną (ObRa, ObRc, ObRd), które uczestniczą, między innymi w transporcie leptyny przez dwie fizjologiczne bariery: bariera krew – mózg i krew – płyn mózgowo-rdzeniowy.
Poza wyżej wymienionymi lokalizacjami obecność receptorów leptynowych wykazano także w komórkach β trzustki, wątrobie, sercu, przednim płacie przysadki, jądrach, jajnikach i w nadnerczach (19, 20, 21, 22, 24, 56).
Podstawową funkcją leptyny jest kontrola bilansu energetycznego ustroju w mechanizmie hamowania łaknienia na poziomie podwzgórza. Poposiłkowy wzrost stężenia leptyny powoduje obniżenie poziomu neuropeptydu Y (NPY), który wykazuje działanie antagonistyczne w stosunku do leptyny (NPY zaliczany jest do peptydów oreksygenicznych – zwiększających łaknienie) (22, 23, 24, 56, 57). Zmniejszona ekspresja genu leptyny i obniżone jej stężenie wykazane zostały w okresie głodzenia i w jadłowstręcie psychicznym ( anorexia nervosa) (58, 59, 60).
Jednakże u osób otyłych stwierdzany jest często podwyższony poziom leptyny spowodowany opornością na leptynę prawdopodobnie w wyniku zaburzonego transportu do ośrodkowego układu nerwowego, jak również zaburzonego mechanizmu przekazywania sygnału (zaburzona funkcja JAK-kinazy i białka STAT3) (61, 62).
Leptyna odgrywa ważną rolę nie tylko w utrzymaniu prawidłowej homeostazy energetycznej ustroju, ale wpływa także na funkcjonowanie innych układów. Wywiera ona modulujący wpływ na układ immunologiczny. Wykazano, że leptyna stymuluje produkcję IL2 i IFNγ przez limfocyty Th1, a hamuje produkcję IL4 przez limfocyty Th2, nasila aktywację i proliferacje monocytów, które są głównym źródłem syntezy TNFα. Badania przeprowadzone u myszy ob./ob. (zwierząt nie posiadających zdolności syntezy leptyny) wykazały zmniejszoną odporność tych zwierząt i większą ich śmiertelność w przebiegu eksperymentalnego modelu sepsy wywołanej lipopolisacharydami (LPS) bakterii Gram ujemnych (63, 64, 65, 66).
Publikacje dotyczące wpływu leptyny na procesy rozwoju miażdżycy wskazują, że leptyna wywołuje wielorakie efekty aterogenne, m.in. wpływając na dysfunkcję śródbłonka, stymulując stres oksydacyjny i reakcję zapalną, zwiększając agregację płytek krwi oraz proliferację komórek mięśniówki gładkiej naczyń (67).
Leptyna poza opisanymi powyżej działaniami odgrywa istotną rolę w procesach rozrodczych, stymulacjąc pulsacyjne wydzielanie GnRH w podwzgórzu i wtórnie syntezę gonadotropin w przysadce (56).
Tumor Necrosis Factor α (TNFα) –czynnik martwicy nowotworu
Jest to jedna z najważniejszych cytokin prozapalnych. W tkance tłuszczowej TNFα wydzielany jest przez adipocyty oraz makrofagi znajdujące się w zrębie. Rola TNFα w procesach metabolicznych obejmuje hamowanie aktywności genów związanych z metabolizmem kwasów tłuszczowych i glukozy, zmniejszanie wydzielania adiponektyny, co pośrednio nasila insulinooporność. TNFα swoje działanie wywiera głównie na poziomie tkankowym. Należy również podkreślić fakt, że stężenia TNFα w surowicy dodatnio korelują z ilością tkanki tłuszczowej (21, 22, 23, 24, 26, 46).
Interleukina 6 (IL6)
IL6, podobnie jak TNFα, jest również cytokiną prozapalną zwiększającą insulinooporność. IL6 i jej receptor wykazują ekspresję w komórkach tkanki tłuszczowej, przy czym ekspresja w wisceralnej tkance tłuszczowej jest 2-3 krotnie silniejsza niż w tkance tłuszczowej podskórnej. Analogicznie do TNFα stężenie IL6 koreluje dodatnio z ilością tkanki tłuszczowej. IL6 hamuje także syntezę adiponektyny. Sugeruje się, że IL6 może być predyktorem cukrzycy typu 2 i chorób układu sercowo-naczyniowego.
Na poziomie ośrodkowego układu nerwowego wpływ IL6 na homeostazę energetyczną ustroju jest odmienny w stosunku do działania obwodowego. Stężenia IL6 w OUN korelują ujemnie z masą tkanki tłuszczowej, co może wskazywać na rolę niedoboru IL6 w OUN w rozwoju otyłości. W badaniach eksperymentalnych podanie IL6 do OUN powoduje u zwierząt doświadczalnych wzrost wydatkowania energii (21, 22, 23, 24, 26, 68).
Rola neuropeptydów wydzielanych w ośrodkowym układzie nerwowym i obwodowo w kontroli łaknienia
Łaknienie regulowane jest przez szereg czynników, zarówno endo-jak i egzogennych. Kontroli podlegają, m.in. wielkość i częstość spożywanych posiłków, wybór rodzaju diety, co konsekwencji prowadzi do kontroli ilości dostarczanej energii. Zaburzenie regulacji łaknienia stanowi istotny czynnik wpływający na rozwój nadwagi i otyłości. Szeroko pojęte zachowania żywieniowe kontrolowane są przez neuropeptydy wydzielane w ośrodkowym układzie nerwowym i przewodzie pokarmowym, adipokiny produkowane w tkance tłuszczowej, jak również pozostają pod wpływem bodźców psychologicznych. Istotne znaczenie w kontroli ilości i jakości przyjmowanych pokarmów odgrywa podwzgórze, w którym zlokalizowane są ośrodki głodu i sytości.
Neuropeptydy zarówno pochodzenia centralnego, jak i obwodowego (powstające głównie w przewodzie pokarmowym i tkance tłuszczowej) zostały podzielone na dwie przeciwstawne grupy: peptydy oreksygeniczne (zwiększające łaknienie) i anoreksygeniczne (hamujące łaknienie). Najważniejszymi peptydami oreksygenicznymi są syntetyzowane w OUN: neuropeptyd Y, oreksyny, Agouti-related peptide (AgRP), galanina, endogenne opioidy, peptydy układu endokanabinoidowego oraz syntetyzowana, przede wszystkim w komórkach żołądka grelina (69, 70, 71). Do drugiej grupy peptydów anoreksygenicznych zalicza się: peptydy ośrodkowe, w tym cocaine-amphetamine regulated transcript (CART), proopiomelanokortynę (POMC), czynnik pobudzający wydzielanie kortykotropiny (CRF), serotoninę, a do wydzielanych w przewodzie pokarmowym cholecystokininę, peptyd YY, amylinę, insulinę, bombezynę oraz syntetyzowaną w tkance tłuszczowej leptynę (69, 70, 71, 72).
Kluczową rolę w mechanizmach kontroli łaknienia odgrywa podwzgórze, w którym w jądrze brzuszno-przyśrodkowym zlokalizowany jest ośrodek sytości, a w jądrach bocznych ośrodek głodu. Natomiast jadro łukowate (nucleus arcuatus) pełni rolę ośrodka integrującego wszystkie sygnały związane z kontrolą łaknienia docierające do układu podwzgórzowego. W jądrze łukowatym zlokalizowane są dwa główne systemy kontroli łaknienia:
? Układ oreksygeniczny – neuropeptyd Y/białko Agouti (AgRP).
? Układ anoreksygeniczny – POMC/CART.
Układy te regulują nie tylko uczucie głodu i sytości, ale także wpływają na wydatkowanie energii i aktywność układu współczulnego (69, 70, 71).
Neuropeptyd Y (NPY) jest polipeptydem zbudowanym z 36 aminokwasów i występuje on w neuronach wykazujących ekspresję białek Agouti. NPY zaliczany jest do bardzo silnych stymulatorów łaknienia, a jego obecność została stwierdzona w licznych komórkach OUN ze szczególnym uwzględnieniem jądra łukowatego. Wywiera on swoje działanie przez 5 typów receptora Y, spośród których receptor Y5 wydaje się mieć największe znaczenie. Ekspresja NPY zmniejsza się pod wpływem, między innymi leptyny i insuliny, a jego aktywność wzrasta w okresie głodzenia, laktacji i cukrzycy z niedoborem insuliny oraz w wyniku wysiłku fizycznego (71, 73, 74).
Agouti related peptide (AgRP) jest 132 aminokwasowym białkiem obecnym tylko w jadrze łukowatym podwzgórza. Neurony wydzielające AgRP posiadają zdolność do syntezy także NPY. Ekspresja AgRP ulega zwiększeniu w przypadku braku leptyny oraz podczas głodzenia. Ponadto białko to jest również selektywnym antagonistą receptorów melanokortyny (MC3 i MC4), co jest jednym z mechanizmów jego działania oreksygenicznego (69, 70, 71, 75, 76).
Cocaine-amphetamine regulated transcript (CART) stanowi grupę peptydów o różnej długości cząsteczek polipeptydowych. Za najaktywniejszy biologicznie uznawany jest fragment CART 55-102. Dotychczas nie udało się znaleźć specyficznych receptorów dla tej grupy peptydów, natomiast wykazano stymulujący wpływ CART na współczulny układ nerwowy, oś podwzgórzowo-przysadkowo-nadnerczową czy wydzielanie hormonów przysadkowych (np. TSH, GH) (77, 78, 79, 80). Neurony wydzielające CART zlokalizowane są w OUN, przede wszystkim w okolicach jąder przykomórowych i jąder bocznych podwzgórza oraz w jądrze łukowatym. W jądrach przykomorowych mRNA dla CART występuje wraz z neuronami wazopresynowymi oraz zawierającymi CRF. Ekspresję CART wykazano też w narządach obwodowych, takich jak trzustka lub nadnercza (77, 78).
Sugeruje się, że mechanizmy działania CART i leptyny mogą ze sobą interferować, ponieważ neurony zawierające CART zlokalizowane są w podwzgórzu, w okolicach bogatych w receptory leptynowe. Poza tym, podanie leptyny myszom ob./ob. zwiększa ekspresję mRNA dla CART. Natomiast CART podawany zdrowym szczurom blokuje zależną od NPY hiperfagię (70, 71, 81).
Na podstawie analizowanego piśmiennictwa można również stwierdzić, że CART wywiera działanie plejotropowe, gdyż poza hamowaniem łaknienia moduluje czynność układu endokrynnego, ośrodkowego i autonomicznego układu nerwowego oraz immunologicznego (82).
Układ melanokortyny
Melanokortyna jest aktywnym biologicznie peptydem powstającym w wyniku potranslacyjnego przekształcania proopiomelanokortyny (POMC). W wyniku defragmentacji POMC powstaje, między innymi ACTH, który przez działanie na receptory MC2 (receptor melanokortynowy 2) odgrywa kluczową rolę w kontroli steroidogenezy nadnerczowej (71). Kolejnym istotnym hormonem powstającym z POMC jest melanokortyna (αMSH), mająca istotne znaczenie, między innymi w kontroli łaknienia. αMSH hamuje łaknienie, działając przez receptory melanokortynowe 4 i 3 (MC4 i MC3) (83, 84).
Naturalnym agonistą receptorów MC4 i MC3 jest melanokortyna, a antagonistą jest Agouti Related Peptide (AgRP) (85, 86).
Liczne badania eksperymentalne wykazywały, że podanie do ośrodkowego układu nerwowego melanokortyny powoduje zahamowanie łaknienia, jak również znosi pobudzenie łaknienia wywołane przez AgRP (87). Jednakże mechanizm wzajemnych oddziaływań pomiędzy układem AgRP a melanokortyną na poziomie ośrodkowego układu nerwowego, a w szczególności jąder podwzgórza, nie jest jednoznacznie wyjaśniony, a wpływ melenokortyny i AgRP na jądra boczne podwzgórza może przebiegać niezależnie (88). Podanie myszom ob./ob. leptyny stymuluje syntezę αMSH, natomiast myszy pozbawione receptora MC4 (knockout MC4-receptor mice) wykazują hiperfagię i otyłość (71). Wpływ melanokortyny na bilans energetyczny ustroju dotyczy nie tylko ośrodkowego układu nerwowego, ale także tkanek obwodowych, ponieważ w adipocytach szczurzych wykazano obecność receptorów melanokortynowych, w tym MC4 (71, 89).
W badaniach klinicznych wykazano, że u części osób otyłych od dzieciństwa może występować czynnościowa mutacja receptora MC4 (90, 91).
Badania eksperymentalne dostarczają dowodów o wzajemnym współdziałaniu pomiędzy neuronami zawierającymi CART i POMC, jak również o współdziałaniu układu POMC i leptyny (71).
Dotychczasowe wyniki badań eksperymentalnych i klinicznych wskazują na bardzo istotną rolę układu POMC/melanokortyna w mechanizmie kontroli energetycznej ustroju. Zaburzenia funkcji tego układu mogą prowadzić do otyłości, co znalazło potwierdzenie w badaniach klinicznych.
Układ endokanabinoidowy
Istotną rolę w kontroli łaknienia odgrywa układ endokanabinoidowy związany z grupą związków chemicznych będących alkaloidami roślinnymi zawartymi między innymi w konopiach indyjskich. Pod koniec lat osiemdziesiątych XX wieku zidentyfikowano receptory kanabinoidowe typu 1 i 2 (CB1 i CB2). Receptory CB1 wykryto w ośrodkowym układzie nerwowym (podwzgórze, nucleus arcuatus, nucleus accumbens, pień mózgu), w autonomicznym układzie nerwowym (n. błędny), w wątrobie, mięśniach szkieletowych, w przewodzie pokarmowym i w tkance tłuszczowej (92).
Układ endokanabinoidowy bierze udział w regulacji energetycznej ustroju przez zwiększanie łaknienia w mechanizmie działania endogennego Δ9 tetrahydrocannabinolu na receptory CB1 i CB2. Ze względu na lokalizację receptorów CB w okolicach podwzgórza bogatych w neurony produkujące NPY, AgRP, CRF czy POMC, wydaje się, że opisywany wzrost łaknienia pod wpływem kanabinoidów spowodowany jest prawdopodobnie w wyniku wzrostu ekspresji i produkcji NPY i spadku aktywności CRF (93, 94). Endokanabinoidy regulują także bilans energetyczny organizmu nie tylko na poziomie OUN, ale również w mechanizmie działania obwodowego, hamując opróżnianie żołądka oraz zwalniając perystaltykę jelit. W tkance tłuszczowej pobudzenie receptora endokanabinoidowego prowadzi do nasilenia adipogenezy, natomiast zastosowanie blokerów receptora endokanabinoidowego powoduje zahamowanie łaknienia i redukcję masy ciała (93, 95, 96).
Peptydy obwodowe
Poza neuropeptydami wywierającymi swoje działanie na poziomie centralnego układu nerwowego istotną rolę odgrywają peptydy syntetyzowane w tkankach obwodowych, przede wszystkim w przewodzie pokarmowym, do których zaliczamy grelinę (peptyd oreksygeniczny) oraz cholecystokininę i peptyd YY (PYY) (peptydy anoreksygeniczne). Grelina jest 28-aminokwasowym peptydem wyizolowanym początkowo z żołądka szczura. Peptyd ten syntetyzowany jest nie tylko w komórkach śluzówki żołądka, ale także w przysadce, podwzgórzu, trzustce, płucach lub układzie immunologicznym (97, 98). Działa przez receptor związany z białkiem G, który zlokalizowany jest, przede wszystkim w przysadce mózgowej. Grelina jest hormonem przewodu pokarmowego, którego poziom wzrasta w czasie głodzenia, co jest bodźcem stymulującym łaknienie. Stymuluje ona wydzielanie NPY i Agouti RP w jadrze łukowatym i w ten sposób działa antagonistyczne w stosunku do leptyny (71, 97, 98, 99). Przedposiłkowy wzrost poziomu greliny w surowicy u ludzi uznawany jest za sygnał do rozpoczęcia posiłku. Pobudzenie receptora greliny (GHS-R) w przysadce powoduje wzrost wydzielania hormonu wzrostu (GH), kortyzolu, katecholamin i prolaktyny. W badaniach klinicznych wykazano podwyższone poziomy greliny u pacjentek z anorexia nervosa (97, 100).
Przedstawione dane wskazują, że grelina odgrywa istotną rolę w kontroli łaknienia zarówno krótkotrwałej, jak i długoterminowej.
Zaburzenia hormonalne w otyłości
Wieloletnie obserwacje i badania potwierdzają, że układ hormonalny włączony jest w skomplikowany system kontroli łaknienia i homeostazy energetycznej ustroju.
Wśród licznych hormonów mających wpływ na kontrolę łaknienia jest insulina. Insulina nasila oksydację glukozy, magazynowanie lipidów i aminokwasów, natomiast hamuje lipolizę, oksydację tłuszczów oraz uwalnianie glukozy zmagazynowanej w postaci glikogenu. Cechą charakterystyczną dużej grupy pacjentów z nadwagą, a przede wszystkim z otyłością jest insulinooporność często połączona z hiperinsulinizmem (101). Analizując zmiany hormonalne w przebiegu otyłości, nie można pominąć faktu wzajemnych wpływów pomiędzy hormonami a adipokinami, takimi jak leptyna czy też adiponektyna. Insulina działa pobudzająco na wydzielanie leptyny i hamuje wydzielanie adiponektyny (14, 22, 23, 102).
Redukcja masy ciała uzyskiwana przy pomocy samej diety, jak też w połączeniu z farmakoterapią lub leczeniem operacyjnym, prowadzi nie tylko do zmniejszenia ilości tkanki tłuszczowej i redukcji masy ciała, ale także do zmniejszenia insulinooporności (103, 104).
Hormony nadnerczowe i układ podwzgórze-przysadka-nadnercza
Glikokortykoidy są hormonami o działaniu diabetogennym. Powodują redystrybucję tkanki tłuszczowej, a jednym z klasycznych objawów zespołu Cushinga jest otyłość wisceralna. Kortyzol nasila utlenianie aminokwasów, rozpad białek, glukoneogenezę i syntezę glukozy w wątrobie (28, 105).
Wzrost poziomu endogennego kortyzolu lub kortykoterapia powoduje, między innymi nasilenie łaknienia. Wzrost łaknienia związany jest z hamującym działaniem kortyzolu na wydzielanie CRF (czynnik uwalniający kortykotropinę) – istotnego hormonu o działaniu anoreksygenicznym) (71). Pomimo tego, że w otyłości prostej stężenia kortyzolu w surowicy oraz test hamowania z deksametazonem są prawidłowe, to dochodzi do nadmiernego metabolizmu kortyzolu w tkance tłuszczowej. Może prowadzić to do podwyższenia dobowego wydzielania 17-hydroksykortykosteroidów (17-OHCS) w moczu przy prawidłowym wydalaniu wolnego kortyzolu w moczu (23, 102, 105).
Zaburzenia osi podwzgórzowo-przysadkowo-nadnerczowej prowadzą do zwiększenia liczby receptorów dla glikokortykoidów w tkance tłuszczowej trzewnej. Wzrost poziomu kortyzolu zwiększa stężenie kwasów tłuszczowych w surowicy oraz nasila gromadzenie się wisceralnej tkanki tłuszczowej, co sprzyja rozwojowi insulinooporności. Istotną rolę w rozwoju insulinooporności, cukrzycy typu 2 oraz zespołu metabolicznego pod wpływem kortyzolu odgrywa także wzrost aktywności 11β hydroksysteroidowej dehydrogenazy typu 1, która przekształca nieaktywny kortyzon w kortyzol. Enzym ten wytwarzany w adipocytach powoduje zwiększenie lokalnej aktywności glikokortykoidów i zwiększenie liczby receptorów dla glikokortykoidów w tkance tłuszczowej (23, 102, 106).
Badania wzajemnych zależności pomiędzy glikokortykoidami a leptyną wykazały hamujący wpływ leptyny na oś podwzgórze-przysadka-nadnercza. Z drugiej strony, glikokortykoidy podwyższają stężenia leptyny (w zespole Cushinga stwierdzana jest hiperleptynamia, co może jednak być spowodowane istniejącym także hiperinsulinizmem) (56, 107, 108).
Zjawisko zwiększonego metabolizmu hormonów nadnerczowych dotyczy także androgenów, w wyniku czego dochodzi do wzrostu ich syntezy, a w konsekwencji u części pacjentów stwierdzane jest zwiększone wydalanie 17 ketosteroidów w moczu dobowym. Sugeruje się, że zaburzenia metabolizmu androgenów mogą wpływać na dystrybucję tkanki trzewnej i nasilać otyłość trzewną (23, 102).
Oś podwzgórzowo-przysadkowo-nadnerczowa odgrywa bardzo istotną rolę w patogenezie otyłości zarówno na poziomie centralnym, jak i obwodowym. Dotychczasowe wyniki badań eksperymentalnych i klinicznych wskazują na konieczność dalszego uściślenia mechanizmów działania glikokortykoidów i androgenów nadnerczowych w otyłości.
Hormon wzrostu
Wydzielanie hormonu wzrostu (hGH) w otyłości jest zaburzone i charakteryzuje się zmniejszeniem wydzielania podstawowego i po stymulacji GHRH (somatoliberyną) lub w wyniku hipoglikemii poinsulinowej (28, 101, 102).
Hormon wzrostu działa anaboliczne poprzez nasilenie syntezy białek oraz redukcję ilości tkanki tłuszczowej w wyniku pobudzenia lipolizy. Otyłość typu brzusznego występuje u pacjentów z niedoborem hormonu wzrostu, natomiast leczenie osób z somatotropową niedoczynnością przysadki powoduje zmniejszenie ilości wisceralnej tkanki tłuszczowej (109, 110).
Próby leczenia otyłości hormonem wzrostu oraz badania doświadczalne wskazują na możliwość pozytywnego efektu hormonu wzrostu w procesie redukcji masy ciała i ilości tkanki tłuszczowej. Mechanizmami korzystnego działania hormonu wzrostu wydają się być, antagonistyczne działanie GH na lipogenetyczny efekt insuliny, jak również hamowanie lipazy lipoproteinowej w komórkach tłuszczowych (111).
Oś podwzgórze-przysadka-tarczyca
Hormony tarczycy wykazują bardzo istotny wpływ na kontrolę metabolizmu ustroju. Zarówno w nadczynności, jak i niedoczynności tarczycy, występują głębokie zaburzenia metaboliczne mające wpływ na funkcjonowanie całego organizmu. W sytuacji ciężkich chorób ogólnoustrojowych (zawał serca, wstrząs, sepsa, urazy wielonarządowe, rozległe zabiegi operacyjne, wyniszczenie) dochodzi do zmian funkcjonowania osi podwzgórze-przysadka-tarczyca określanych jako „euthyroid sick syndrome”.
Wzrost masy ciała w przypadku niedoczynności tarczycy czy utrata masy ciała w nadczynności wskazują na potencjalne istotne znaczenie hormonów tarczycy w patogenezie otyłości. Wiadomo jest, że hipertyreoza nasila insulinooporność w mechanizmie zaburzeń postreceptorowych oraz poprzez stymulację glukoneogenezy (23). Jednakże badania oceniające wpływ leczenia subklinicznej niedoczynności tarczycy nie wykazują istotnego wpływu hormonów tarczycy na redukcję masy ciała, natomiast w przypadku pełnoobjawowej niedoczynności tarczycy z istotnymi zaburzeniami hormonalnymi suplementacja l-tyroksyny prowadzi do redukcji masy ciała na poziomie ok. 10% (112).
Badania dotyczące wzajemnych korelacji pomiędzy leptyną a czynnością osi podwzgórze-przysadka-tarczyca wskazują na pobudzający wpływ leptyny na wydzielanie TRH. Ponadto hormony tarczycy i leptyna nasilają termogenezę, zwiększając wydatek energetyczny przez stymulację białek rozprzęgających (UCP – uncoupled protein) znajdujących się głównie w brunatnej tkance tłuszczowej i mięśniach (113, 114).
Powyższe omówienie podstawowych zaburzeń endokrynnych w przebiegu otyłości nie wyczerpuje oczywiście całego złożonego zagadnienia. Przedstawione dane wydają się mieć najistotniejsze znaczenie w zaburzeniach endokrynologicznych w otyłości. Należy pamiętać, że hormony, takie jak katecholaminy, serotonina, GLP-1, glukagon, IGF-1 mogą w sposób istotny modyfikować stan metaboliczny i energetyczny ustroju.
Wydaje się, że wiele opisanych zaburzeń w wydzielaniu neuropeptydów i hormonów ma istotny związek z hiperinsulinizmem i insulinoopornością, co w konsekwencji prowadzić może do rozwoju zespołu metabolicznego ze wszelkimi jego powikłaniami.
Czynniki genetyczne otyłości
Otyłość związana jest z wieloma czynnikami genetycznymi i środowiskowymi. Badania rodzin osób otyłych wykazują, że czynniki genetyczne wpływają na BMI w 30-40% (dotyczy to pokrewieństwa rodzice-dzieci i rodzeństwo-rodzeństwo), natomiast oddziaływania czynników środowiskowych (takich jak skład diety, sposób przyjmowania pokarmów, wzorce rodzinne i kulturowe odżywiania) wynoszą ok. 60-70% (115). Jednakże w szeregu opublikowanych badań wskazuje się na dużo bardziej istotną rolę czynników genetycznych w patogenezie otyłości. W badaniach tych, dotyczących bliźniąt jedno- i dwujajowych, określa się w przybliżeniu, że czynniki genetyczne odpowiedzialne są w 70%, a nawet w 90%, choć szacunki te wydają się być zawyżone (115, 116, 117, 118).
Czynniki genetyczne bez wątpienia warunkują predyspozycje do wystąpienia otyłości, jednakże określanie otyłości jako choroby uwarunkowanej genetycznie wydaje się mieć ograniczone uzasadnienie, za czym przemawia kilka faktów:
1. Lawinowo narastająca ilość osób otyłych na świecie, w szczególności w krajach wysokorozwiniętych, spowodowana jest, przede wszystkim zmianami w rodzaju stosowanej diety i zmniejszeniu aktywności fizycznej, a nie w zmianie genów występujących w danych populacjach.
2. Wśród populacji Indian Pima zamieszkujących południowo-zachodnie regiony Stanów Zjednoczonych (rezerwat w Arizonie) stwierdzany jest jeden z najwyższych na świecie odsetek występowania otyłości i cukrzycy typu 2, natomiast u Indian Pima żyjących w Meksyku otyłość występuje sporadycznie.
3. Pomimo stwierdzanych pomiędzy poszczególnymi populacjami różnic w występowaniu otyłości sugerujących istotną rolę czynników genetycznych, migracja i związane z nią zmiany w sposobie odżywania i trybie życia prowadzą najczęściej do znacznego wzrostu częstości występowania otyłości.
4. Modyfikacja czynników środowiskowych, a w szczególności diety, prowadzi nie tylko do redukcji masy ciała i BMI, ale także do zmniejszenia ryzyka wystąpienia chorób współistniejących z otyłością (101).
Otyłość wydaje się być zatem problemem klinicznym uwarunkowanym wieloczynnikowo i stanowi wynik oddziaływania pomiędzy predyspozycją genetyczną do rozwoju otyłości a obesogenicznymi czynnikami środowiskowymi. Już w latach 60-tych ubiegłego stulecia sformułowana została teoria tzw. oszczędzającego genotypu, który miał być ewolucyjnym przystosowaniem się człowieka do cyklicznie występujących okresów głodu (119).
Ustalenie wzajemnych relacji gen-czynniki środowiskowe oraz ich realnego wpływu na rozwój otyłości jest istotnym problemem, a rozwiązanie tych zagadnień wymaga licznych, dalszych badań (101, 120).
Pod względem uwarunkowań genetycznych z otyłości można wydzielić choroby dziedziczone jednogenowo lub wielogenowo. Jednogenowe mutacje prowadzące z reguły do bardzo zaawansowanych stadiów otyłości występują bardzo rzadko i nie stanowią one istotnego problemu epidemiologicznego (121). Wydaje się, że dziedziczenie wielogenowe ma zasadnicze znaczenie w występowaniu predyspozycji do otyłości (115, 116, 117, 118).
W patogenezie otyłości najlepiej poznanymi mutacjami pojedynczego genu są mutacje: genu receptora melanokortyny 4, mutacje genów leptyny (model zwierzęcy – szczep myszy ob./ob.) lub genu receptora leptyny (szczep myszy db/db) (115, 116, 121).
W ostatnim czasie opublikowano szereg doniesień dotyczących polimorfizmu genu FTO (FAT mass and obesity-associated gene) jako istotnego czynnika w rozwoju otyłości. Badania prowadzone w Europie wskazują, że wśród dzieci i młodzieży obecność polimorfizmu AA (rs9939609) w intronie tego genu prowadzić może do wzrostu masy ciała związanego z większą ilością przyjmowanych pokarmów i słabiej wyrażonym poposiłkowym uczuciem sytości (badania brytyjskie) (122, 123). Jednakże badania autorów niemieckich dotyczące tego samego polimorfizmu genu FTO u dzieci i młodzieży z obszaru Niemiec nie wykazały zmian w zakresie wskaźników lipidowych, glukozy i BMI-SDS w przypadku obecności allelu AA (124). Stwierdzono również, że mRNA powstający na matrycy DNA genu FTO wykazuje wysoką ekspresję w podwzgórzu w rejonach odpowiedzialnych za kontrolę łaknienia. Udowodniono, że ekspresja genu FTO w jądrze łukowatym podwzgórza jest zależna od ilości przyjmowanego pokarmu (122, 125). Inne polimorfizmy genu FTO (rs8050136) związane są z większą masą ciała u badanych osób, co spowodowane jest ilością spożywanych kalorii, a nie zmniejszonym wydatkiem energetycznym (126). Powyższe dane wskazują na konieczność prowadzenia dalszych badań w tym zakresie w celu uzyskania bardziej jednoznacznych wyników.
Innym czynnikiem genetycznym, który wydaje się także mieć znaczenie w rozwoju otyłości, jest gen receptora aktywowanego proliferatorem peroksysomów, kodujący receptor PPARγ. Receptory te odgrywają bardzo ważną rolę w mechanizmie metabolizmu lipidów w zależności od dostępności tłuszczów dla organizmu. Stwierdzono, że ekspresja genu tego receptora, jak również samego receptora, może być modulowana przez czynniki dietetyczne. Badania eksperymentalne u myszy wykazały, że głodzone zwierzęta miały obniżoną ekspresję receptora PPARγ, natomiast dieta wysokotłuszczowa powodowała zwiększenie ekspresji receptorów PPARγ w komórkach tłuszczowych (127). Badania polimorfizmów tego receptora u ludzi wykazały, że występowanie polimorfizmu pro12ala może wiązać się ze zmniejszonym ryzykiem cukrzycy typu 2, zwiększoną wrażliwością na insulinę i niższym BMI (128).
Opisane powyżej przykłady dotyczące zarówno genu FTO, jak i PPARG, potwierdzają konieczność analizy czynników genetycznych w otyłości w odniesieniu do czynników środowiskowych, takich jak rodzaj diety czy łaknienie.
Wiadomo także, że tkanka tłuszczowa jest nie tylko magazynem energetycznym ustroju, ale także istotnym organem wydzielania wewnętrznego. Wydzielane przez nią peptydy, hormony czy enzymy mają istotne znaczenie w kontroli łaknienia (np. leptyna) czy metabolizmie ogólnoustrojowym (m.in. adiponektyna, rezystyna, wisfatyna). Zaburzenia genetyczne związane z syntezą lub działaniem tych peptydów mogą być związane z predyspozycjami do wystąpienia otyłości.
Do szczególnie istotnych zaliczyć należy mutacje genu leptyny i receptora leptyny prowadzące do niemożności syntezy tego peptydu lub nieprawidłowego działania receptora leptynowego. Inne opisywane zmiany dotyczą genów insulinopodobnego czynnika 1 i 2, lipazy lipoproteinowej, neuropeptydu Y i jego receptora typu 5 oraz receptora PPARγ (115, 116, 117, 128). Wśród potencjalnie istotnych zaburzeń rolę odgrywają polimorfizmy związane z genem adiponektyny. Są to zmiany pojedynczego nukleotydu w regionie promotorowym SNP 11 426 (A/G), SNP 11 391 (G/A), SNP 11 377 (C/G), zmiany w genie powodujące przyłączenie glicyny w pozycji 15 polipeptydu, tj. w sekwencji sygnałowej, która to sekwencja nie występuje w dojrzałym białku (SNP 45 (T/G). Polimorfizmy mogą też dotyczyć zmian w obrębie intronów (SNP 276 G/T) lub zmian w obrębie tzw. domeny włóknistej lub globularnej cząsteczki adiponektyny (31, 129, 130, 131).
Wymienione powyżej zmiany o podłożu genetycznym związane mogą być z większym prawdopodobieństwem wystąpienia cukrzycy typu 2, insulinooporności oraz nadciśnienia tętniczego. Uznaje się, że omówione uprzednio polimorfzmy genu adiponektyny mogą także zwiększać ryzyko wystąpienia zespołu metabolicznego (131). Wyniki badań polimorfizmów genu adiponektyny prowadzących do zmian struktury białka lub możliwości tworzenia trimerów, heksamerów lub większych kompleksów nie są jednoznaczne. Uzyskiwane wyniki wykazują rozbieżności w zależności od badanej populacji (31, 129, 130, 131, 132, 133).
Rezystyna jest adipokiną związaną prawdopodobnie z rozwojem insulinooporności w przebiegu otyłości i zespołu metabolicznego. Wyższe stężenia rezystyny w osoczu i surowicy stwierdzono u chorych z cukrzycą i otyłością (43, 134). Do najczęściej opisywanych w literaturze polimorfizmów rezystyny należy polimorfizm -420C/G zlokalizowany w części promotorowej genu. Polimorfizm ten związany jest z występowaniem otyłości, insulinooporności i cukrzycy typu 2 w różnych badanych populacjach. Polimorfizm +62G/A wydaje się korelować z otyłością, jednakże dane dotyczące jego związku z ryzykiem wystąpienia cukrzycy są niejednoznaczne (43, 135). W badaniach niemieckich obecność polimorfizmu +62G/A wiązała się z obecnością nadciśnienia tętniczego, ale nie cukrzycy (136).
Wyniki badań dotyczących związku polimorfizmów genu rezystyny z występowaniem cukrzycy typu 2, otyłości, nadciśnienia tętniczego, a w konsekwencji zespołu metabolicznego, są niejednoznaczne i wymagają dalszych szczegółowych opracowań.
UZASADNIENIE CELOWOŚCI PODJĘCIA BADAŃ
Dotychczas opublikowano szereg danych na temat otyłości i zespołu metabolicznego pochodzących z licznych badań eksperymentalnych i klinicznych, ale pomimo to, nadal istnieje wiele niejasności dotyczących patofizjologii, jak i skutków zdrowotnych związanych z otyłoscią i zespołem metabolicznym.
Wraz z rozwojem stopnia zaawansowania klinicznego otyłości mierzonego przy pomocy BMI stwierdzany jest stopniowy wzrost częstości występowania zespołu metabolicznego. Jednakże nawet u pacjentów z otyłością olbrzymią (BMI ≥ 40 kg/m2) zespół metaboliczny nie jest rozpoznany u wszystkich chorych. Od kilku lat w publikacjach ta szczególna grupa chorych, bez zespołu metabolicznego, określana jest jako otyli metabolicznie zdrowi. Zatem, jeżeli pomimo istnienia tak zawansowanej klinicznie otyłości pacjenci ci nie mają nasilonych zaburzeń metabolicznych, czy też nie stwierdza się u nich cukrzycy typu 2 lub nadciśnienia tętniczego, to istotnym wydaje się być pytanie, czy pomiędzy pacjentami otyłymi z lub bez zespołu metabolicznego istnieją różnice w wydzielaniu adipokin oraz czy ewentualnie tym zmianom towarzyszą asocjacje genetyczne. Dotychczas u tych pacjentów nie wykazano jednoznacznych zmian w profilu wydzielanych adipokin lub też odmienności w zakresie wskaźników genetycznych mających związek z wystąpieniem lub brakiem obecności zespołu metabolicznego.
W niniejszej pracy została podjęta próba oceny czynników genetycznych z zastosowaniem analizy częstości występowania polimorfizmów genów dwóch adipokin o przeciwstawnych działaniach: adiponektyny o działaniu zmniejszającym insulinoooporność, posiadającej właściwości przeciwmiażdżycowe i antydiabetogenne oraz rezystyny, która uznawana jest za adipokinę stymulującą rozwój insulinooporności. Wykazanie istotnych statystycznie różnic w występowaniu poszczególnych genotypów badanych polimorfzmów lub ich asocjacji z zespołem metabolicznym mogłoby stać się potencjalnym predyktorem ewentualnych zaburzeń metabolicznych w otyłości.
Połączenie badań dotyczących zmian stężenia adipokin w różnych grupach kobiet z nadwagą i otyłością w powiązaniu z oceną insulinooporności wraz z badaniami polimorfizmów genów adiponektyny i rezystyny może pozwolić na wyodrębnienie grupy pacjentek o szczególnie nasilonych zmianach metabolicznych, które to pacjentki powinny być poddane intensywnemu nadzorowi medycznemu. Ważnym punktem tego badania jest także fakt, że wszystkie badania dotyczące oceny stopnia klinicznego zaawansowania otyłości i oceny insulinooporności oraz gospodarki lipidowej są możliwe do wykonania w warunkach ambulatoryjnych i nie wymagają specjalistycznej aparatury badawczej, co pozwala na ich szerokie stosowanie w warunkach pozaszpitalnych zarówno w gabinetach specjalistycznych, jak i przez lekarzy pierwszego kontaktu.
CEL PRACY
Celem pracy jest wyodrębnienie czynników wpływających na wystąpienie zespołu metabolicznego u osób z nadwagą i otyłością.
Analizie zostaną poddane zmiany stężeń adipokin i peptydów związanych z kontrolą łaknienia w grupach pacjentek z obecnością lub brakiem zespołu metabolicznego z różnym stopniem zaawansowania otyłości i nadwagi.
Oceniane będą także korelacje pomiędzy analizowanymi peptydami a wskaźnikami isnulinooporności, wynikami badań parametrów lipidowych i parametrami antropometrycznymi.
Badane będą także asocjacje polimorfizmów genów adiponektyny i rezystyny w odniesieniu do obecności zespołu metabolicznego u pacjentek z nadwagą lub otyłością.
Na podstawie przedstawionych powyżej założeń badawczych podjęta zostanie próba określenia ewentualnych markerów peptydowych lub genetycznych pozwalających na wyodrębnienie grupy pacjentek o podwyższonym ryzyku wystąpienia zespołu metabolicznego.
ZADANIA BADAWCZE
1. Ocena parametrów antropometrycznych, wskaźników gospodarki lipidowej i węglowodanowej w wybranej grupie kobiet oraz kwalifikacja ich do poszczególnych grup badanych:
a) pacjentki szczupłe, zdrowe z BMI <25 kg/m2,
b) pacjentki z nadwagą i otyłością z BMI 25-39,9 kg/m2 skojarzoną z zespołem metabolicznym,
c) pacjentki z nadwagą i otyłością z BMI 25-39,9 kg/m2 bez zespołu metabolicznego,
d) pacjentki z otyłością olbrzymią z BMI ≥ 40 kg/m2.
2. Ocena zmian stężeń adipokin i peptydów (leptyny, adiponektyny całkowitej i wysokocząsteczkowej, wisfatyny, greliny, rezystyny) w poszczególnych badanych grupach pacjentek z nadwagą i otyłością w stosunku do grupy kontrolnej, jak również porównanie pomiędzy poszczególnymi wymienionymi powyżej badanymi grupami pacjentek.
3. Ocena zmian stężeń peptydów, adipokin i wyników badań biochemicznych pomiędzy pacjentkami z obecnością lub brakiem zespołu metabolicznego.
4. Ocena korelacji i zależności zmian stężeń adipokin w stosunku do wskaźników antropometrycznych, gospodarki lipidowej i węglowodanowej w poszczególnych badanych grupach kobiet z nadwagą i otyłością.
5. Ocena zmian częstości występowania badanych polimorfizmów genów adiponektyny i rezystyny oraz ocena ilorazu szans pomiędzy następującymi grupami kobiet:
a) pacjentki z nadwagą i otyłością skojarzoną z zespołem metabolicznym w stosunku do kobiet z nadwagą i otyłością bez zespołu metabolicznego,
b) pacjentki z nadwagą i otyłością skojarzoną z zespołem metabolicznym w stosunku do kobiet z prawidłowym BMI,
c) pacjentki z nadwagą i otyłością bez zespołu metabolicznego w stosunku do kobiet z prawidłowym BMI.
6. Próba określenia asocjacji pomiędzy zespołem metabolicznym a analizowanymi polimorfizmami.
7. Próba znalezienia markerów peptydowych i/lub genetycznych różnicujących nadwagę i otyłość skojarzoną z zespołem metabolicznym w stosunku do nadwagi i otyłości bez zespołu metabolicznego.
MATERIAŁ I METODY
Badania przeprowadzono łącznie u 369 kobiet, które podzielono na następujące grupy: 34 kobiety z otyłością olbrzymią (BMI ≥ 40 kg/m2), 100 kobiet z nadwagą i otyłością i zespołem metabolicznym (BMI 25-39,9 kg/m2), 131 kobiet z nadwagą i otyłością (BMI 25-39,9 kg/m2) bez zespołu metabolicznego. Grupę kontrolną stanowiły 104 młode kobiety z prawidłową masą ciała (BMI 18-24,9 kg/m2). Wszystkie badane pacjentki były włączone do badania w trybie ambulatoryjnym.
Zespół metaboliczny rozpoznawano na podstawie kryteriów sformułowanych przez International Diabetic Federation (IDF 2005).
Nadciśnienie tętnicze definiowano wg kryteriów ESH i ESC, gdy wartości RR wynosiły ≥ 140/90 mm Hg lub gdy było ono rozpoznane już wcześniej i pacjentka otrzymywała leczenie hipotensyjne, natomiast cukrzycę typu 2 rozpoznawano zgodnie z następującymi kryteriami: w przypadku glikemii na czczo ≥ 126 mg/dl lub gdy 2 godziny po podaniu 75 g glukozy glikemia ≥ 200 mg/dl lub kiedykolwiek w ciągu doby glikemia wynosiła ≥ 200 mg/dl w osoczu krwi żylnej. Klasyfikację cukrzycy oceniano wg zaleceń Komitetu Ekspertów WHO sformułowanych w 1998 r.
Kryteria wykluczające z włączenia do badań były następujące:
1. choroby układu endokrynnego,
2. przewlekłe choroby układu oddechowego,
3. przewlekłe choroby wątroby i nerek,
4. choroby nowotworowe.
Żadna z badanych kobiet w trakcie przeprowadzonych badań nie wykazywała objawów ostrej choroby zakaźnej, jak również nie otrzymywała leków przeciwzapalnych, nie nadużywała alkoholu i nie paliła papierosów.
Protokół badań został zaakceptowany przez Komisję Bioetyczną w Centrum Medycznym Kształcenia Podyplomowego. Wszystkie osoby badane wyraziły świadomą zgodę na uczestniczenie w badaniu klinicznym i pobranie krwi.
Badanie kliniczne
Pomiar ciśnienia tętniczego krwi wykonano po 15 minutowym odpoczynku w pozycji siedzącej. Wzrost i wagę oraz BMI oceniano wg standardowych procedur. Obwód talii i wskaźnik talia-biodra (waist-hip ratio WHR) stosowano jako wskaźnik otyłości centralnej. Zawartość tkanki tłuszczowej oceniano metodą bioelektrycznej bioimpedancji (BIA) przy pomocy aparatu firmy Tanita.
Insulinooporność mierzono przy pomocy wskaźnika HOMA-IR (homeostasis model assesment metod), który definiowano jako: stężenie insuliny na czczo (?U/ml) X stężenie glukozy na czczo (mmol/l)/22,5.
Pomiar stężenia peptydów i hormonów w osoczu krwi obwodowej oraz ocena parametrów lipidowych i gospodarki węglowodanowej
Krew do badań pobierano rano, na czczo, po 12 godzinach od ostatniego posiłku. Bezpośrednio po pobraniu materiału próbki krwi wirowano w temperaturze +4°C i rozdzielano do 1,5 ml probówek, które przechowywano w temperaturze -70°C. Do probówek przed wirowaniem dodawano inhibitory proteaz, aprotyninę i EDTA. Próbki przeznaczone do oznaczeń peptydowych nie podlegały wcześniejszemu rozmrożeniu lub naprzemiennemu rozmrożeniu/zamrożeniu.
Profil lipidowy oraz oznaczenia poziomu glukozy wykonywano wg rutynowych standardowych procedur laboratoryjnych.
Stężenia leptyny, adiponektyny i aktywnej formy greliny w osoczu wykonano metodą radioimmunologiczną (RIA) przy pomocy zestawów firmy Linco Research o następującej czułości: 0,5 ng/ml dla leptyny, 1 ng/ml dla adiponektyny i 7,8 pg/ml dla greliny active.
Poziomy rezystyny i rozpuszczalnego receptora dla leptyny oceniano, stosując testy ELISA firmy Bio Vendor Laboratory Medicine. Czułość testów wynosiła odpowiednio 0,1 ng/ml dla rezystyny i 1,2 ng/ml dla rozpuszczalnego receptora leptytowego.
Do oznaczenia stężenia C-końcowego fragmentu wisfatyny stosowano metodę EIA (zestawy firmy Phoenix Pharmaceutical INC). Czułość testu wynosiła 0,1 ng/ml.
Insulinę oznaczano metodą IRMA zestawami firmy BioSource (czułość 1 ?U/ml).
Frakcję wysokocząsteczkową adiponektyny (HMW adiponectin) oceniono przy pomocy testu EIA firmy ALPCO o czułości 0,075 ng/ml.
Oznaczenia w/w adipokin i peptydów wykonywane były wg protokołów dołączonych do zestawów.
Zmienność wewnątrzseryjna i międzyseryjna wszystkich stosowanych do oznaczeń zestawów była mniejsza niż 10%.
Badania genetyczne – oznaczanie polimorfizmów pojedynczych nukleotydów dla genu rezystyny i adiponektyny
Genomowe DNA było izolowane z krwi pełnej z dodatkiem EDTA przy pomocy zestawu NucleoSpin Blood Quick Pure Kit (Macherey-Nagel – Niemcy) według załączonego do zestawu protokołu.
Oznaczano następujące polimorfizmy pojedynczego nukleotydu (SNP) dla genu rezystyny: 420 C/G, 62 G/A, 537 A/C oraz następujące SNP dla genu adiponektyny: 45 T/G, 276 G/T, 11 377 C/G, 11 391 G/A. Genotypowanie wykonano przy pomocy TaqMan SNP Genotyping Assays. Reakcja PCR czasu rzeczywistego (Real Time PCR – RT-PCR) była wykonana aparatem ABI PRISM Sequence Detection System (SDS) (Applied Biosystems; Foster City, CA) w 96-dołkowych płytkach. Do każdego dołka dodawano 20 ng genomowego DNA, 1,25 ?l TaqMan SNP genotyping Assay, 12,5 ?l TaqMan Universal Master Mix (Applied Biosystems; Foster City, CA) i 9,25 ?l wody. W każdej płytce 2 dołki nie zawierały genomowego DNA. Amplifikację DNA uzyskiwano wg następującej procedury: 1 – cykl 95°C przez 10 min, a następnie 40 cykli wg schematu 92°C przez 15 sek. i 60°C przez 1 min. Rozdział poszczególnych alleli w zakresie kolejnych polimorfizmów wykonywano aparatem ABI PRISM 7000 SDS Software.
ANALIZA STATYSTYCZNA
Analizę statystyczną wykonano przy użyciu pakietu STATISTICA ver 7.1 PL, ustalając poziom istotności statystycznej α = 0,05.
Normalność rozkładu w obrębie poszczególnych grup badano testami: Shapiro-Wilka oraz Kołmogorowa-Smirnowa z poprawką Lillieforsa.
Ze względu na brak normalności rozkładu danych w części badanych parametrów do weryfikacji hipotezy dotyczącej zróżnicowania więcej niż 2 grup stosowano test Kruskala-Wallisa (nieparametryczny odpowiednik analizy wariancji), a przy porównaniu dwóch grup stosowano test Manna-Whitneya (nieparametryczny odpowiednik testu t-Studenta dla prób niezależnych).
Korelację pomiędzy badanymi adipokinami: (leptyna, adiponektyna, HMW adiponektyna, rezystyna, wisfatyna) i peptydami (Grelin active) oraz wynikami badań antropometrycznych i badań biochemicznych oceniano przy użyciu korelacji Spearmana (nieparametryczny odpowiednik korelacji Pearsona).
Po zbadaniu założeń wykonano analizę kowariancji ANCOVA w badanych grupach, gdzie jako zmienną zależną analizowano adiponektynę całkowitą i wysokocząsteczkową, leptynę, rezystynę, wisfatynę i grelinę, a jako zmienną wikłającą uznano kolejno: BMI, BIA i WHR.
Wyniki badań peptydów, adipokin, wskaźników antropometrycznych i lipidowych (dane ilościowe) zostały przedstawione jako średnia ± odchylenie standardowe (SD).
Porównania częstotliwości dystrybucji genotypów poszczególnych analizowanych polimorfizmów genów adiponektyny i rezystny wykonano odpowiednio przy pomocy testu x2 (w zależności od spełnienia założeń tego testu stosowano wersję standardową lub z poprawką Yates´a albo dokładny test Fishera).
Iloraz szans (OR) wraz z 95% przedziałem ufności (CI) oceniano przy pomocy kalkulatora dostępnego na http://www.hutchon.net/ConfidOR.htm. Ocenę tę przeprowadzono dla modelu dominującego, który definiowano jako heterozygoty + homozygoty recesywne vs homozygoty dominujące.
Oceniano także zgodność rozkładu genotypów poszczególnych badanych polimorfizmów z prawem Hardyego-Wienberga w grupie kontrolnej.
W celu oceny asocjacji analizowanych polimorfizmów genów adiponektyny i rezystyny z nadwagą, otyłością i zespołem metabolicznym oraz jednoczesnej oceny ilorazu szans pomiędzy poszczególnymi grupami pacjentek wykonano analizę regresji logistycznej w następujących układach:
1. Pacjentki z otyłością i nadwagą skojarzoną z zespołem metabolicznym w odniesieniu do kobiet z nadwagą i otyłością bez towarzyszącego zespołu metabolicznego.
2. Pacjentki z nadwagą i otyłością z zespołem metabolicznym w stosunku do grupy kobiet młodych z prawidłowym BMI.
3. Pacjentki z nadwagą i otyłością bez zespołu metabolicznego w porównaniu do grupy młodych kobiet z prawidłowym BMI.
WYNIKI
Tabela 2 A. Liczebność grup pacjentek z otyłością olbrzymią oraz nadwagą i otyłością po podzieleniu tych pacjentek w zależności od obecności lub braku zespołu metabolicznego.
Tabela 2 B. Częstość występowania zespołu metabolicznego wśród badanych pacjentek.
Tabela 2 C. Częstość występowania nadciśnienia tętniczego i cukrzycy wśród badanych pacjentek.
Tabela 3. Analiza danych klinicznych pacjentek z BMI ≥ 25 kg/m2 (wszystkie analizowane grupy z nadwagą i otyłością) w stosunku do grupy kontrolnej.
Dane przedstawione jako średnia ± SD
Tabela 4. Wartości wskaźników biochemicznych i stężeń peptydów u pacjentek z z BMI ≥ 25 kg/m2 (wszystkie analizowane grupy z nadwagą i otyłością) w stosunku do grupy kontrolnej.
Dane przedstawione jako średnia ± SD
Tabela 5. Analiza danych klinicznych pacjentek z BMI ≥ 25 kg/m2 podzielonych na grupy z obecnością zespołu metabolicznego ZM (+) lub bez zespołu metabolicznego ZM (-) w stosunku do grupy kontrolnej.
Dane przedstawione jako średnia ± SD
BMI> 25 kg/m 2 ZM (-)– otyłość i nadwaga bez zespołu metabolicznego
BMI> 25 kg/m 2 ZM (+)– otyłość i nadwaga z zespołem metabolicznym a – BMI> 25 kg/m 2 ZM (-) vs BMI> 25 kg/m 2 ZM (+) b – BMI> 25 kg/m 2 ZM (-) vs grupa kontrolna c – BMI> 25 kg/m 2 ZM (+) vs grupa kontrolna Tabela 6. Wartości wskaźników biochemicznych i stężeń peptydów u pacjentek z BMI ≥ 25 kg/m2 podzielonych na grupy z lub bez obecności zespołu metabolicznego oraz pacjentek z grupy kontrolnej.
Dane przedstawione jako średnia ± SD
BMI> 25 kg/m 2 ZM (-)– otyłość i nadwaga bez zespołu metabolicznego
BMI> 25 kg/m 2 ZM (+)– otyłość i nadwaga z zespołem metabolicznym a – BMI> 25 kg/m 2 ZM (-) vs BMI> 25 kg/m 2 ZM (+) b – BMI> 25 kg/m 2 ZM (-) vs grupa kontrolna c – BMI> 25 kg/m 2 ZM (+) vs grupa kontrolna Tabela 7. Dane kliniczne pacjentek z otyłością olbrzymią (BMI ≥ 40 kg/m2), otyłością i nadwagą bez zespołu metabolicznego (BMI 25,00-39,9 kg/m2), otyłością i nadwagą z zespołem metabolicznym (BMI 25,00-39,9 kg/m2) w stosunku do grupy kontrolnej.
Wyniki przedstawione jako średnia ± SD
***p <0,001 vs grupa kontrolna
Tabela 8. Wartości wskaźników biochemicznych i stężeń peptydów pacjentek z otyłością olbrzymią (BMI ≥ 40 kg/m2), otyłością i nadwagą bez zespołu metabolicznego (BMI 25,00-39,9 kg/m2), otyłością i nadwagą z zespołem metabolicznym (BMI 25,00-39,9 kg/m2) w stosunku do grupy kontrolnej.
Wyniki przedstawione jako średnia ± SD
***p <0,001 vs grupa kontrolna; **p<0,01; vs grupa kontrolna; *p<0,05 vs grupa kontrolna
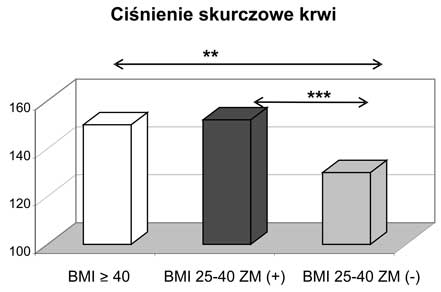 Ryc. 1. Wartości skurczowego ciśnienia tętniczego u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001; **p <0,01
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 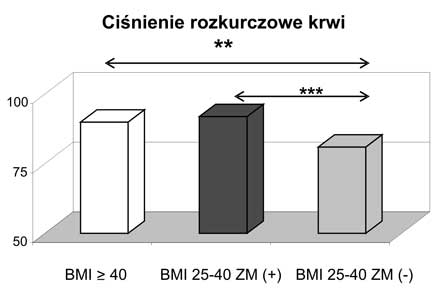 Ryc. 2. Wartości rozkurczowego ciśnienia tętniczego u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001; **p <0,01
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 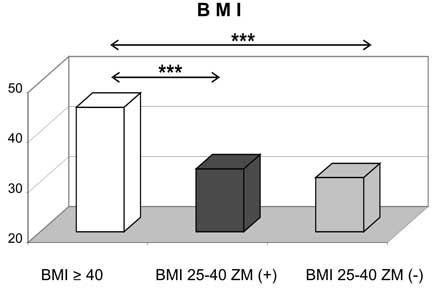 Ryc. 3. Wartości BMI u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001; **p <0,01
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 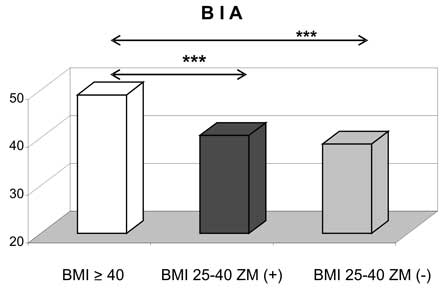 Ryc. 4. Wartości BIA u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 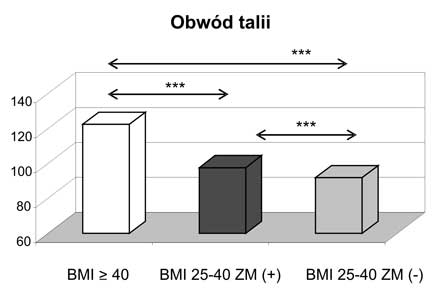 Ryc. 5. Wartości obwodu talii u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 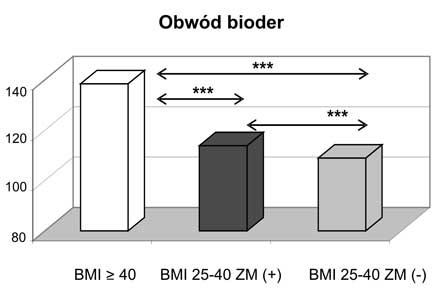 Ryc. 6. Wartości obwodu bioder u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 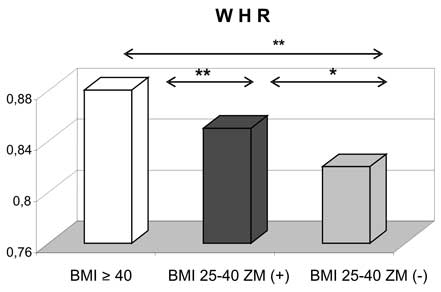 Ryc. 7. Wartości wskaźnika talia-biodra (WHR) u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
**p <0,01; *p<0,05
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 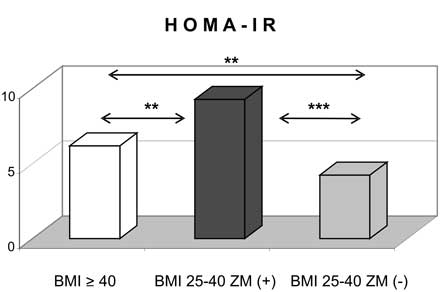 Ryc. 8. Wartości HOMA-IR u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001; **p <0,01
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 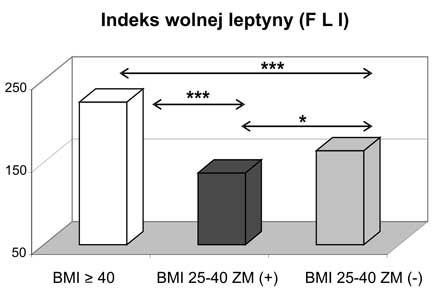 Ryc. 9. Wartości Indeksu wolnej leptyny (FLI) u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001; **p <0,01; *p <0,05
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 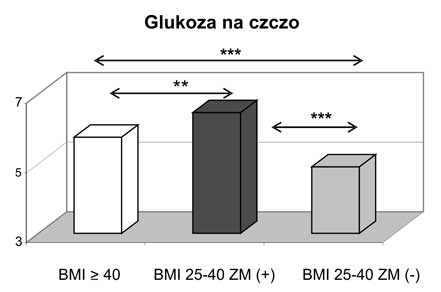 Ryc. 10. Wartości glukozy na czczo w surowicy u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001; **p <0,01; *p <0,05
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 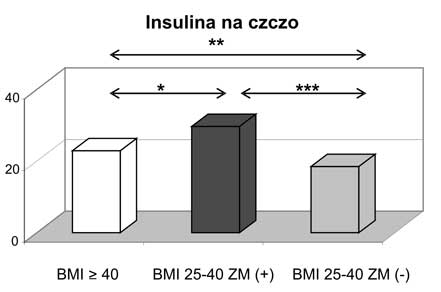 Ryc. 11. Wartości insuliny na czczo w surowicy u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001; **p <0,01; *p <0,05
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 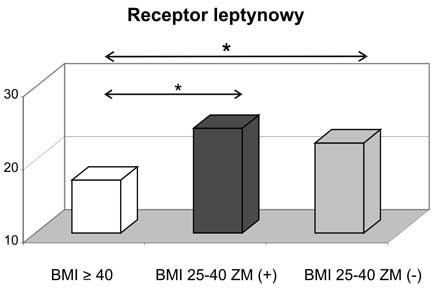 Ryc. 12. Stężenie receptora leptynowego w surowicy u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
*p <0,05
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 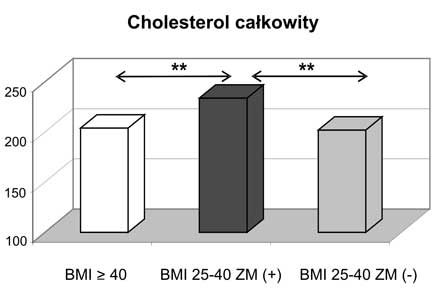 Ryc. 13. Stężenia cholesterolu całkowitego w surowicy u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
**p <0,01
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 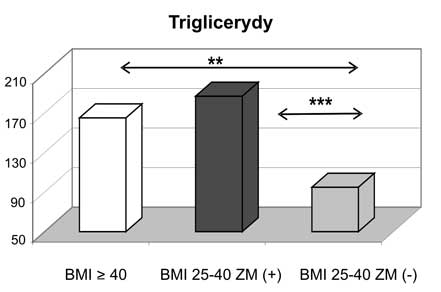 Ryc. 14. Stężenia triglicerydów w surowicy u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001; **p <0,01
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 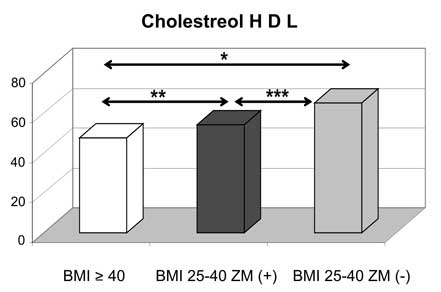 Ryc. 15. Stężenia HDL-cholesterolu w surowicy u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001; **p <0,01; *p <0,05
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 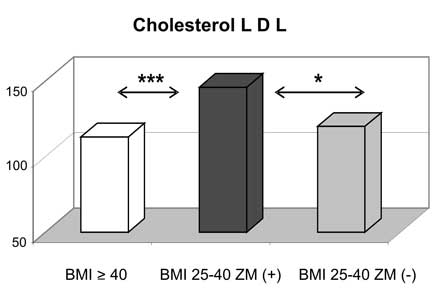 Ryc. 16. Stężenia LDL-cholesterolu w surowicy u pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001; *p <0,05
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+) – nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 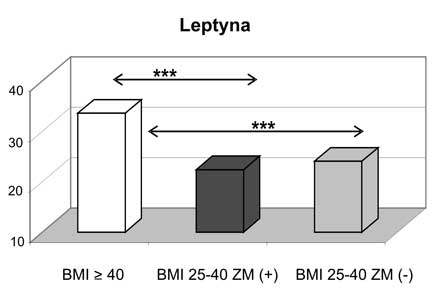 Ryc. 17. Stężenia leptyny w surowicy pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 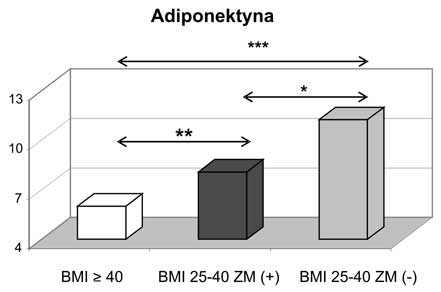 Ryc. 18. Stężenia adiponektyny całkowitej w surowicy pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001; **p <0,01; *p <0,05
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 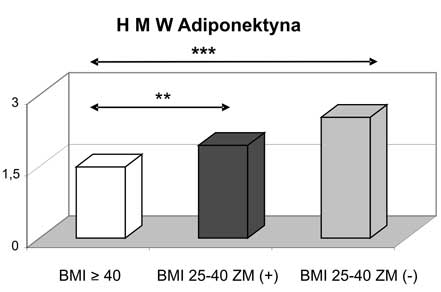 Ryc. 19. Stężenia wysokocząsteczkowej adiponektyny w surowicy pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
***p <0,001; **p <0,01
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 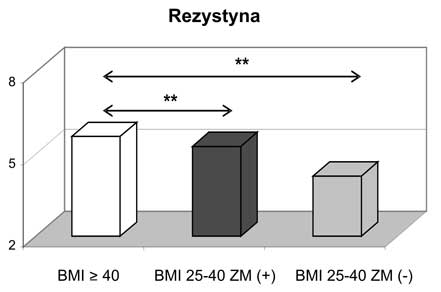 Ryc. 20. Stężenia rezystyny w surowicy pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
**p <0,01
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. 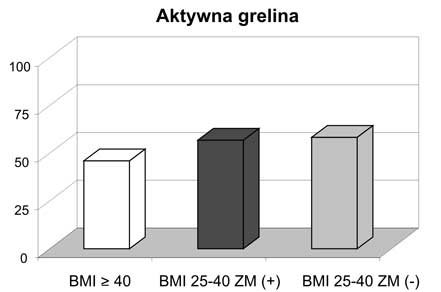 Ryc. 21. Stężenia aktywnej greliny w surowicy pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym.
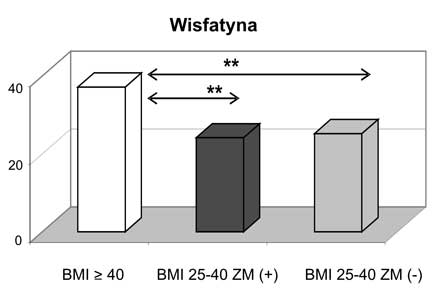 Ryc. 22. Stężenia wisfatyny w surowicy pacjentek z poszczególnych badanych grup kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym lub bez zespołu metabolicznego.
**p <0,01
BMI ≥ 40 – otyłość olbrzymia; BMI 25-40 ZM (-)– nadwaga i otyłość z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; BMI 25-40 ZM (+)– nadwaga i otyłość z BMI 25-39,9 kg/m2 z zespołem metabolicznym. Tabela 9. Analiza danych klinicznych pacjentek z otyłością olbrzymią podzielonych na 2 podgrupy:
1. Otyłość olbrzymia (BMI ≥ 40 kg/m2) z towarzyszącym zespołem metabolicznym N = 25 kobiet 2. Otyłość olbrzymia (BMI ≥ 40 kg/m2) bez towarzyszącego zespołu metabolicznego N = 9 kobiet wraz z porównaniem do grupy kontrolnej Dane przedstawione jako średnia ± SD
***p<0,001; **p<0,01; *p<0,05
OLB ZM (-) – otyłość olbrzymia bez zespołu metabolicznego OLB ZM (+) – otyłość olbrzymia z zespołem metabolicznym a – OLB ZM (-) vs OLB ZM (+) b – OLB ZM (-) vs grupa kontrolna c – OLB ZM (+) vs grupa kontrolna Tabela 10. Wartości wskaźników biochemicznych i stężeń peptydów u pacjentek z otyłością olbrzymią podzielonych na 2 podgrupy wraz z porównaniem do grupy kontrolnej.
1. Otyłość olbrzymia (BMI ≥ 40 kg/m2) z towarzyszącym zespołem metabolicznym N = 25 kobiet OLB ZM (+) 2. Otyłość olbrzymia (BMI ≥ 40 kg/m2) bez towarzyszącego zespołu metabolicznego N = 9 kobiet OLB ZM (-) Dane przedstawione jako średnia ± SD
OLB ZM (-) – otyłość olbrzymia bez zespołu metabolicznego
OLB ZM (+) – otyłość olbrzymia z zespołem metabolicznym a – OLB ZM (-) vs OLB ZM (+) b – OLB ZM (-) vs grupa kontrolna c – OLB ZM (+) vs grupa kontrolna Tabela 11. Znamienne statystycznie korelacje badanych neuropeptydów z parametrami uzyskanymi w badaniu przedmiotowym dla całej grupy pacjentek z BMI ≥ 25 (nadwaga i otyłość bez zespołu metabolicznego, nadwaga i otyłość skojarzona z zespołem metabolicznym, otyłość olbrzymia).
Tabela 12. Znamienne statystycznie korelacje badanych neuropeptydów z parametrami metabolicznymi dla całej grupy pacjentek z BMI ≥ 25 (nadwaga i otyłość bez zespołu metabolicznego, nadwaga i otyłość skojarzona z zespołem metabolicznym, otyłość olbrzymia).
Tabela 13. Znamienne statystycznie korelacje badanych neuropeptydów z parametrami uzyskanymi w badaniu przedmiotowym dla grupy pacjentek z BMI ≥ 40 (otyłość olbrzymia).
Tabela 14. Znamienne statystycznie korelacje badanych neuropeptydów z parametrami metabolicznymi dla grupy pacjentek z BMI ≥ 40 (otyłość olbrzymia).
Tabela 15. Znamienne statystycznie korelacje badanych neuropeptydów z parametrami uzyskanymi w badaniu przedmiotowym dla grupy pacjentek z nadwagą i otyłością (BMI 25-39,9 kg/m2) i zespołem metabolicznym.
Tabela 16. Znamienne statystycznie korelacje badanych neuropeptydów z parametrami metabolicznymi dla grupy pacjentek z nadwagą i otyłością (BMI 25-39,9 kg/m2) i zespołem metabolicznym.
Tabela 17. Znamienne statystycznie korelacje badanych neuropeptydów z parametrami uzyskanymi w badaniu przedmiotowym dla grupy pacjentek z nadwagą i otyłością (BMI 25-39,9 kg/m2) bez zespołu metabolicznego.
Tabela 18. Znamienne statystycznie korelacje badanych neuropeptydów z parametrami metabolicznymi dla grupy pacjentek z nadwagą i otyłością (BMI 25-39,9 kg/m2) bez zespołu metabolicznego.
Tabela 19. Rozkład genotypów dla poszczególnych badanych polimorfizmów u pacjentek z otyłością i nadwagą (BMI ≥ 25 kg/m2) z towarzyszącym zespołem metabolicznym w porównaniu do kobiet z otyłością i nadwagą (BMI ≥ 25 kg/m2) bez zespołu metabolicznego.
Tabela 20. OR i 95% CI w modelu dominującym dla poszczególnych analizowanych polimorfizmów u pacjentek z otyłością i nadwagą (BMI ≥ 25 kg/m2) oraz z zespołem metabolicznym w stosunku do kobiet z nadwagą i otyłością (BMI ≥ 25 kg/m2) bez zespołu metabolicznego.
Tabela 21. Rozkład genotypów dla poszczególnych badanych polimorfizmów u pacjentek z otyłością i nadwagą (BMI ≥ 25 kg/m2) z towarzyszącym zespołem metabolicznym w porównaniu do kobiet szczupłych.
Rozkład genotypów poszczególnych badanych polimorfizmów był zgodny z prawem Hardyego-Wienberga w grupie kontrolnej.
Tabela 22. Rozkład genotypów dla poszczególnych badanych polimorfizmów u pacjentek z otyłością i nadwagą (BMI ≥ 25 kg/m2) bez towarzyszącego zespołu metabolicznego w porównaniu do kobiet szczupłych.
Rozkład genotypów poszczególnych badanych polimorfizmów był zgodny z prawem Hardyego-Wienberga w grupie kontrolnej.
Tabela 23. OR i 95% CI w modelu dominującym dla poszczególnych analizowanych polimorfizmów u pacjentek z otyłością olbrzymią w stosunku do grupy kontrolnej.
Rozkład genotypów poszczególnych badanych polimorfizmów był zgodny z prawem Hardyego-Wienberga w grupie kontrolnej.
Tabela 24. OR i 95% CI w modelu dominującym dla poszczególnych analizowanych polimorfizmów u pacjentek z otyłością i nadwagą (BMI ≥ 25 kg/m2) oraz z zespołem metabolicznym w stosunku do grupy kontrolnej.
Rozkład genotypów poszczególnych badanych polimorfizmów był zgodny z prawem Hardyego-Wienberga w grupie kontrolnej.
Tabela 25. OR i 95% CI w modelu dominującym dla poszczególnych analizowanych polimorfizmów u pacjentek z otyłością i nadwagą (BMI ≥ 25 kg/m2) bez zespołu metabolicznego w stosunku do grupy kontrolnej.
Rozkład genotypów poszczególnych badanych polimorfizmów był zgodny z prawem Hardyego-Wienberga w grupie kontrolnej.
Najwyższe wartości ciśnienia tętniczego zanotowano u pacjentek z otyłością olbrzymią oraz nadwagą i otyłością skojarzoną z zespołem metabolicznym.
Najwyższe wartości rozkurczowego ciśnienia tętniczego zanotowano u pacjentek z otyłością olbrzymią oraz nadwagą i otyłością skojarzoną z zespołem metabolicznym.
Wśród analizowanych grup najwyższe wartości BMI stwierdzono u pacjentek z otyłością olbrzymią. Nie zanotowano istotnych statystycznie różnic pomiędzy kobietami z otyłością skojarzoną z zespołem metabolicznym oraz otyłością bez zespołu metabolicznego.
Najwyższe wartości BIA zanotowano u kobiet z otyłością olbrzymią. Wartości BIA w grupach nadwagi i otyłości z towarzyszącym zespołem metabolicznym oraz nadwagi i otyłości bez zespołu metabolicznego nie wykazywały istotnych statystycznie różnic.
W analizowanych grupach najwyższe wartość obwodu talii stwierdzono u kobiet z otyłością olbrzymią i kobiet z nadwagą i otyłością z zespołem metabolicznym. Pacjentki z otyłością i nadwagą bez zespołu metabolicznego miały znamiennie mniejszy obwód talii niż pacjentki z zespołem metabolicznym i BMI 25-39,9 kg/m2.
Najwyższe wartości obwodu bioder zanotowano u pacjentek z BMI ≥ 40 kg/m2 oraz w grupie kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym.
Najwyższe wartości zanotowano u pacjentek z otyłością olbrzymią oraz w grupie nadwagi i otyłości skojarzonej z zespołem metabolicznym. Pacjentki z nadwagą i otyłością bez zespołu metabolicznego miały wskaźnik WHR istotnie statystycznie niższy niż kobiety z otyłością olbrzymią oraz nadwagą i otyłością z zespołem metabolicznym.
HOMA-IR w analizowanych grupach pacjentek była najwyższa w grupie kobiet z nadwagą i otyłością z towarzyszącym zespołem metabolicznym. Wartość ta u pacjentek z nadwagą i otyłością z zespołem metabolicznym była znamiennie statystycznie wyższa niż w grupie otyłości olbrzymiej.
W badanych grupach stwierdzono najwyższe wartości FLI w grupie otyłości olbrzymiej. Wartości uzyskane w tej grupie były znamiennie statystycznie wyższe w stosunku do pozostałych analizowanych grup. Pacjentki z nadwagą i otyłością skojarzoną z zespołem metabolicznym miały znamiennie niższy FLI w stosunku do kobiet z grupy BMI 25-39,9 bez zespołu metabolicznego.
Najwyższe wartości glukozy na czczo stwierdzono u pacjentek z nadwagą i otyłością z zespołem metabolicznym.
Najwyższe wartości insuliny na czczo zanotowano w grupie pacjentek z nadwagą i otyłością skojarzoną z zespołem metabolicznym. Najniższe stężenia insuliny na czczo zanotowano u pacjentek z nadwagą i otyłością bez zespołu metabolicznego.
Pacjentki z otyłością olbrzymią miały znamiennie statystycznie niższe stężenia receptora leptynowego w stosunku do pozostałych badanych grup.
Najwyższe stężenia cholesterolu całkowitego stwierdzono u kobiet z nadwagą i otyłością z zespołem metabolicznym. Wartości te były znamiennie wyższe w stosunku do wszystkich pozostałych badanych grup.
Najwyższe stężenia triglicerydów w surowicy stwierdzono u kobiet z grupy otyłości olbrzymiej oraz nadwagi i otyłości skojarzonej z zespołem metabolicznym. Zanotowano statystycznie znamienne różnice pomiedzy grupami z nadwagą i otyłością skojarzoną lub bez zespołu metabolicznego.
Najwyższe wartości HDL-cholesterolu zanotowano u pacjentek z nadwagą i otyłością bez zespołu metabolicznego. Wartości te były znamiennie wyższe w stosunku do pozostałych badanych grup.
Najwyższe stężenia LDL-cholesterolu zanotowano u kobiet z nadwagą i otyłością oraz zespołem metabolicznym. Uzyskane w tej grupie wartości były znamiennie statystycznie wyższe w stosunku do pozostałych analizowanych grup.
Uzyskane wartości leptyny były najwyższe u pacjentek z otyłością olbrzymią. Nie zanotowano istotnych statystycznie różnic pomiędzy pacjentkami z nadwagą i otyłością skojarzoną z zespołem metabolicznym i bez zespołu metabolicznego.
Najniższe wartości adiponektyny obserwowano w grupie kobiet z otyłością olbrzymią oraz u pacjentek z nadwagą i otyłością z towarzyszącym zespołem metabolicznym. Wykazano znamiennie statystycznie niższe wartości adiponektyny u pacjentek z nadwagą i otyłością z towarzyszacym zespołem metabolicznym w stosunku do kobiet z nadwagą i otyłością bez zespołu metabolicznego.
Uzyskane wartości HMW adiponektyny były najniższe u pacjentek z otyłością olbrzymią. Istotne statystycznie różnice pomiędzy analizowanymi grupami zostały przedstawione na wykresie. Pacjentki z nadwagą i otyłością z zespołem metabolicznym miały poziomy HMW adiponektyny niższe niż pacjentki z nadwagą i otyłością bez zespołu metabolicznego, jednakże różnice te nie były istotne statystycznie.
Najwyższe stężenia rezystyny stwierdzono u kobiet z otyłością olbrzymią oraz nadwagą i otyłością skojarzoną z zespołem metabolicznym. Nie zanotowano zamniennych różnic w stężeniach rezystyny pomiędzy pacjentkami z nadwagą i otyłością z lub bez zespołu metabolicznego.
Nie zanotowano istotnych statystycznie różnic pomiędzy analizowanymi grupami pacjentek z otyłością i nadwagą.
Stwierdzane najwyższe wartości wisfatyny u pacjentek z otyłością olbrzymią były istotnie statystycznie wyższe w stosunku do pozostałych analizowanych grup. Nie zanotowano znamiennych różnic w stężeniach wisfatyny pomiędzy pacjentkami z nadwagą i otyłością z towarzyszącym lub bez zespołu metabolicznego.
Analiza kowariancji (Ancova)
Przeprowadzona analiza kowariancji ANCOVA dla zmiennych zależnych adiponektyny całkowitej i wysokocząsteczkowej, leptyny, rezystyny, wisfatyny i greliny przy kolejno badanych zmiennych wikłających: BMI, BIA i WHR nie wykazała różnic w uzyskanych powyżej wynikach.
Wyniki regresji logistycznej: nie stwierdzono znamiennego statystycznie modelu regresji logistycznej w przypadku porównania (zmienna zależna Y w modelu):
? grupy kobiet z otyłością i nadwagą skojarzoną z zespołem metabolicznym (Y = 1) i grupy kobiet z otyłością i nadwagą bez zespołu metabolicznego (Y = 0) w odniesieniu do analizowanych polimorfizmów (zmienna X).
Suplement wyników badań polimorfizmów genów adiponektyny i rezystyny.
Poniższe wyniki zostają przedstawione jako suplement ze względu na to, że grupa kontrolna młodych szczupłych kobiet różni się pod względem wieku od grup kobiet z nadwagą i otyłością z lub bez zespołu metabolicznego. Trudno jest zatem ocenić, u których kobiet z grupy kontrolnej wystąpi w przyszłości nadwaga lub otyłość.
Wyniki regresji logistycznej.
Znamienny statystycznie model regresji logistycznej stwierdzono w przypadku porównania (zmienna zależna Y w modelu):
? grupy kobiet z otyłością i nadwagą skojarzoną z zespołem metabolicznym (Y = 1) w stosunku do młodych kobiet z prawidłowym BMI (Y = 0)
Natomiast istotnymi czynnikami w tym modelu różnicującymi powyższe grupy pacjentek (zmienna X) były poniższe polimorfizmy genów adiponektyny i rezystyny:
? SNP genu adiponektyny 11 377 C/G OR 2,64 95% CI (1,14-6,10) p<0,05
? SNP genu adiponektyny 276 G/T OR 2,78 95% CI (1,21-6,43) p<0,05
? SNP genu rezystyny 420 C/G OR 2,32 95% CI (1,06-5,11) p<0,05
Poziom istotności statystycznej dla całego modelu p <0,01.
W pozostałych układach wymienionych w rozdziale „Materiał i metody” nie stwierdzono istotnych statystycznie modeli regresji logistycznej.
DYSKUSJA I OMÓWIENIE UZYSKANYCH WYNIKÓW
Analiza uzyskanych wyników zostanie przeprowadzona w oparciu o podział kobiet badanych na podgrupy w zależności od wskaźnika BMI oraz obecności zespołu metabolicznego.
Grupa kobiet z BMI ≥ 25 kg/m2 (otyłość olbrzymia, nadwaga i otyłość z lub bez zespołu metabolicznego – analiza łączna wszystkich grup)
W badanym materiale osoby z nadwagą i otyłością w stosunku do grupy kobiet z prawidłową masą ciała wykazywały istotne statystycznie odrębności parametrów uzyskanych w badaniu przedmiotowym (wyższe wartości BMI, BIA, obwodu talii i bioder, WHR, ciśnienia tętniczego skurczowego i rozkurczowego). Ocena profilu lipidowego u kobiet z nadwagą i otyłością umożliwiła wykazanie zaburzeń lipidowych pod postacią podwyższonych poziomów cholesterolu całkowitego, LDL-cholesterolu i triglicerydów oraz obniżonego stężenia HDL-cholesterolu. Jednakże należy podkreślić, iż w przypadku stężeń triglicerydów średnia wartość uzyskana we wszystkich analizowanych grupach łącznie wynosiła 140 mg/dl i była wartością niższą niż uznawana obecnie wartość 150 mg/dl będąca jednym z kryteriów rozpoznania zespołu metabolicznego. Zatem można stwierdzić, iż obecność nadmiernej ilości tkanki tłuszczowej nie skutkuje bezpośrednio i w każdym przypadku hipertriglicerydemią. Jednocześnie analizując ww. wyniki trzeba uwzględnić fakt, że spośród wszystkich analizowanych osób z BMI ≥ 25 kg/m (łącznie 265 kobiet), tylko u 125 (47%) rozpoznano zespół metaboliczny, a wartości triglicerydów> 150 mg/dl są jednym z kryteriów rozpoznania zespołu metabolicznego. Z piśmiennictwa wynika, że opisywane zaburzenia lipidowe pod postacią, przede wszystkim obniżonego poziomu HDL-cholesterolu i podwyższonego stężenia LDL-cholesterolu wiążą się ze wzrostem ilości wolnych kwasów tłuszczowych i białka transportującego estry cholesterolu – cholesterol ester transfer protein (CETP). Wiadomo, że wzrost poziomu wolnych kwasów tłuszczowych wpływa bezpośrednio na rozwój insulinoooporności, natomiast wzrost poziomu CETP skutkuje obniżeniem HDL-cholesterolu (68). Liczne badania populacyjne wskazują, że obniżenie poziomu HDL-cholesterolu jest jednym z głównych czynników rozwoju choroby niedokrwiennej serca. Zaburzenia lipidowe stwierdzane w otyłości są również zależne od podwyższonych poziomów glikemii lub współistnienia cukrzycy. Hiperglikemia powoduje nasilenie procesów oksydacyjnych lipoprotein LDL. Cząsteczki LDL poddane modyfikacji, w tym oksydacji oraz glikacji, wychwytywane są przez makrofagi, które tworzą komórki piankowate odgrywające ważną rolę w tworzeniu blaszki miażdżycowej. Ponadto zmienione w procesie oksydacji cząsteczki LDL mogą wykazywać szereg niekorzystnych działań, m.in. mogą wpływać na syntezę czynników wzrostowych i cytokin, pobudzać chemotaksję monocytów i makrofagów, zmniejszać produkcję tlenku azotu (NO) w śródbłonku naczyniowym, zaburzając w ten sposób rozkurcz małych naczyń, jak również mogą działać cytotoksycznie na mięśniówkę ściany naczynia i nasilać w tych mechanizmach procesy aterogenezy. Zaburzenia lipidowe stanowią istotny element wpływający na powikłania ogólnoustrojowe w przebiegu otyłości i zespołu metabolicznego (21, 68, 137, 138).
Wraz z rozwojem otyłości dochodzi do stopniowego powiększania się wymiarów komórek tłuszczowych, których wielkość nierzadko przekracza 100 μm. Powoduje to pogorszenie ukrwienia adipocytów i stopniowy rozwój hipoksji, co powodować może martwicę części adipocytów (21, 57, 68, 139). Wynikiem tych procesów patologicznych jest wzrost chemotaksji makrofagów i monocytów do podścieliska tkanki tłuszczowej. W podścielisku tkanki tłuszczowej stwierdzana jest obecność dwóch typów makrofagów: typ M1 dominujący w otyłości, syntetyzujący cytokiny prozapalne (m.in. IL6, TNFα) oraz makrofagi typu M2 syntetyzujące cytokiny przeciwzapalne (m.in. IL10) (140, 141). Udokumentowane oddziaływania parakrynne pomiędzy makrofagami a adipocytami mogą prowadzić do rozwoju insulinooporności i przewlekłego stanu zapalnego w przebiegu otyłości. Poza tym TNFα wydzielany w nadmiernych ilościach u otyłych osób nasila oporność na insulinę i stymuluje produkcję IL6, ICAM-1 czy białka ostrej fazy (CRP), będących typowymi mediatorami stanu zapalnego (142, 143).
Istotną rolę w rozwoju przewlekłego zapalenia i rozwoju insulinooporności wydaje się mieć, oprócz TNFα, również IL6. W badaniach eksperymentalnych IL6 podana obwodowo nasila insulinooporność i stymuluje wydzielanie VEGF (vascular endothelial growth factor), który pobudza angiogenezę w czasie zwiększania ilości i wielkości komórek tkanki tłuszczowej (144, 145). Wiadomo także, że infiltracja makrofagów produkujących IL6 jest większa w tkance tłuszczowej trzewnej niż podskórnej. Jednocześnie w trzewnej tkance tłuszczowej osób otyłych syntetyzowana jest większość cytokin prozapalnych. Wydaje się, że przewlekły proces zapalny spowodowany wzrostem ilości makrofagów w tkance tłuszczowej oraz zwiększoną produkcją cytokin prozapalnych, prowadzi do dysfunkcji tkanki tłuszczowej jako narządu wydzielania wewnętrznego i skutkuje istotnymi zaburzeniami w syntezie adipokin (21, 34, 57, 68). Zatem, wzrost ilości tkanki tłuszczowej występujący w otyłości powoduje zmiany w wydzielaniu licznych adipokin, które odgrywają istotną rolę metaboliczną oraz wpływają na rozwój powikłań metabolicznych otyłości.
Leptyna
Wzrost stężenia leptyny wraz ze zwiększaniem się ilości tkanki tłuszczowej w organizmie jest charakterystycznym zjawiskiem u osób z nadwagą i otyłością. Dane uzyskane z obecnego badania, w tym wyniki badania przedmiotowego przeprowadzonego u pacjentek z BMI ≥ 25 kg/m2, wykazały ścisłą korelację pomiędzy stężeniem leptyny w surowicy krwi a ilością tkanki tłuszczowej (potwierdzeniem jest dodatnia korelacja pomiędzy leptyną a BMI, BIA, obwodem talii i bioder) oraz wskazały na istotną zależność między poziomem leptyny a insulinoopornością (o czym świadczą dodatnie korelacje leptyny ze stężeniem insuliny i HOMA-IR). Poza tym wykazana ujemna korelacja tego peptydu z HDL-cholestrerolem może także wskazywać na znaczenie leptyny w patogenezie miażdżycy.
Udowodniono, że wzrost stężenia leptyny w surowicy w nadwadze i otyłości związany jest nie tylko ze wzrostem ilości i wielkości adipocytów, ale także ze zwiększoną syntezą cytokin prozapalnych, które pobudzają syntezę leptyny. Dodatkowo, pobudzająco na syntezę leptyny wpływają także insulina i wolne kwasy tłuszczowe, których poziom jest podwyższony u osób otyłych (21, 22, 67, 146).
Hiperleptynemia wydaje się być związana ze zwiększonym ryzykiem rozwoju miażdżycy, jednakże nie jest do końca wyjaśnione, czy proaterogenne obwodowe działanie leptyny zależy bezpośrednio od podwyższonego stężenia leptyny w surowicy czy też jest skutkiem oporności na leptynę na poziomie ośrodkowego układu nerwowego (przede wszystkim w podwzgórzu) (62, 67).
Innymi proaterogennymi działaniami tego peptydu jest jej stymulacyjny wpływ na powstawanie wolnych rodników, działanie prozapalne oraz stymulacja akumulacji estrów cholesterolu przez komórki piankowate, które, jak wspomniano wcześniej, mają istotne znaczenie w rozwoju blaszki miażdżycowej (67, 147).
Istotne znaczenie dla rozwoju powikłań sercowo-naczyniowych w nadwadze i otyłości ma wpływ leptyny na dysfunkcję środbłonka naczyniowego. Większość badań in vivo wskazuje na niekorzystne oddziaływanie leptyny na wydzielanie tlenku azotu (NO) przez komórki endotelialne. Postuluje się, że zwiększone stężenia leptyny, które występują w otyłości, wywołują zaburzenia zależnego od NO rozkurczu mięśni gładkich naczyń (148, 149). Jednakże w warunkach in vitro, w badaniach wykonanych na izolowanych tętnicach wieńcowych wykazano, że bardzo wysokie stężenia leptyny mogą wywierać działania wazorelaksacyjne (150, 151). Prospektywne badania wieloośrodkowe wskazują, że wartości leptyny mogą być predyktorem incydentów sercowo-naczyniowych, jednakże nie do końca jest to jednoznacznie udowodnione (67). Sugeruje się również, że wyższe poziomy leptyny mogą być powiązane z epizodami udarów niedokrwiennych i krwotocznych ośrodkowego układu nerwowego (152).
Adiponektyna
W uzyskanych wynikach stwierdzono znamienne obniżenie adiponektyny w grupie kobiet z BMI ≥ 25 kg/m2 w stosunku do grupy kontrolnej.
Adiponektyna korelowała dodatnio z HDL-cholestrerolem, a ujemnie ze skurczowym i rozkurczowym ciśnieniem tętniczym, BMI, BIA, obwodem talii, WHR, HOMA-IR i triglicerydami.
Uzyskane wyniki badań wskazują na przeciwmiażdżycowy wpływ adiponektyny, o czym świadczą, między innymi ujemne korelacje adiponektyny ze wskaźnikami insulinooporności, triglicerydami oraz dodatnia korelacja z HDL-cholesterolem.
Dane z piśmiennictwa potwierdzają, że synteza adiponektyny jest obniżona u osób z otyłością, insulinoopornoscią, zespołem metabolicznym i cukrzycą typu 2. Szereg badań udowadnia, że adiponektyna poprawia insulinowrażliwość oraz wywiera działanie antyaterogenne. Mechanizmy tych działań polegają na zahamowaniu glukoneogenezy wątrobowej i zwiększeniu utylizacji glukozy przez wątrobę oraz komórki mięśniowe, jak również na nasileniu oksydacji kwasów tłuszczowych i ich metabolizowania w wątrobie i mięśniach (24, 57, 153, 154).
Nie można pominąć także tak istotnej cechy adiponektyny, jaką jest jej działanie przeciwzapalne. Peptyd ten działa supresyjnie na aktywność makrofagów zrębu tkanki tłuszczowej przez zahamowanie TLR-4 (toll like receptor) zlokalizowanego na komórkach immunologicznie kompetentnych, hamując w ten sposób kompleks NF-κB w jądrach komórek układu immunologicznego, który jest odpowiedzialny za syntezę, miedzy innymi cytokin prozapalnych (68, 153).
Poza wyżej wymienionymi właściwościami adiponektyna wykazuje również właściwości wazoprotekcyjne w mechanizmie stymulacji produkcji tlenku azotu przez endotelium, jak również przez modulację ekspresji molekuł adhezyjnych i zmiataczy wolnych rodników (155, 156).
Dokładna rola adiponektyny w chorobach układu sercowo-naczyniowego nie jest dotychczas jednoznacznie określona. Wysokie poziomy adiponektyny stwierdzane w badaniach prospektywnych są predyktorem niskiego ryzyka wystąpienia zawału serca u mężczyzn i choroby niedokrwiennej serca u osób z cukrzycą (157, 158). Jednakże ostatnio opublikowane badania wskazują, że u osób z przewlekłą niewydolnością krążenia, pacjentów z potwierdzoną w koronarografii chorobą niedokrwienną serca lub u chorych z przewlekłą niewydolnością nerek, wysokie wartości adiponektyny mogą wskazywać na większe ryzyko zgonu (35, 36).
Powyższe rozbieżności oraz dokładny mechanizm opisywanych zjawisk nie został dotychczas jednoznacznie wyjaśniony. Część autorów sugeruje, że przyczyną dodatniej korelacji pomiędzy stężeniem adiponektyny a ryzykiem wystąpienia choroby wieńcowej oraz zgonu może być, m.in. oporność na adiponektynę w przebiegu chorób układu krążenia, zaburzenia funkcji nerek i wtórnie obniżony klirens nerkowy adiponektyny lub być może jest to reakcja kompensacyjna na nieme niedokrwienie mięśnia sercowego (35, 36, 37). Prawdopodobnym jest również, że w niewydolności krążenia dochodzi do syntezy adiponektyny w kardiomiocytach pod wpływem czynników natriuretycznych, w tym BDNF (brain derieved natriuretic factor) i współdziałania tych dwóch czynników, co pośrednio potwierdza dodatnia korelacja pomiędzy adiponektyną a BDNF stwierdzana u osób z kacheksją w przebiegu niewydolności serca (159).
W dostępnym piśmiennictwie pojawia się koncepcja dotycząca wzajemnego oddziaływania pomiędzy leptyną a adiponektyną w zakresie regulacji łaknienia. Sugeruje się, że podczas głodzenia adiponektyna stymuluje przez receptor AdipoR1 w podwzgórzu przyjmowanie pokarmów w mechanizmie zwiększenie aktywności kinazy białkowej aktywowanej przez AMP (AMPK), jak również wpływa na zmniejszenie wydatku energetycznego (146, 160).
W analizowanym materiale własnym u kobiet z BMI ≥ 25 kg m2 nie wykazano korelacji pomiędzy stężeniami w surowicy adiponektyny i leptyny. Pomimo iż na temat leptyny i adiponektyny opublikowanych zostało wiele prac, to rola tych adipokin w stanach fizjologii i patologii wydaje się być nie do końca wyjaśniona.
Wysokocząsteczkowa adiponektyna (HMW – adiponektyna)
Stężenia adiponektyny wysokocząsteczkowej (HMW adiponektyna) wśród badanych pacjentek z BMI ≥ 25 kg/m2 były niższe niż u kobiet szczupłych z grupy kontrolnej, jednakże wartości te nie były znamienne statystycznie.
Adiponektyna, jak przedstawiono we wstępie, występuje w kilku oligomerycznych formach. Wydaje się, że forma 18-to merowa adiponektyny tzw. wysokocząsteczkowa (HMW – high molecular weight) jest odpowiedzialna za zmniejszenie insulinooporności, natomiast formy o mniejszej masie cząsteczkowej (tzn. trimery i heksamery) działają, przede wszystkim na poziomie ośrodkowego układu nerwowego (24, 31, 57, 153).
Liczne prace wskazują, że adiponektyna wysokocząsteczkowa wykazuje największą aktywność biologiczną i jest odpowiedzialna za korzystny wpływ na procesy metaboliczne ustroju związane ze wzrostem insulinowrażliwości oraz ochronnym działaniem w stosunku do ryzyka wystąpienia cukrzycy typu 2. Istnieją doniesienia, że mutacje domeny kolagenowej adiponektyny (G84R i G90S) powodują obniżenie stężeń adiponektyny wysokocząsteczkowej i wzrost ryzyka zachorowania na cukrzycę typu 2 (161, 162).
W badaniach własnych u kobiet z BMI ≥ 25 kg/m2 wykazano silną ujemną korelację HMW-adiponektyny ze wskaźnikiem HOMA-IR i poziomem insuliny, a przedstawione wyniki są zgodne z danymi publikowanymi przez innych autorów (24, 31, 33, 34, 153). Dodatkowo, potwierdzeniem ujemnych korelacji insulinooporności i poziomu adiponektyny wysokocząsteczkowej są podwyższone poziomy tej adipokiny w surowicy krwi obwodowej u osób z zespołem Pradera-Willego, w którym zanotowano relatywnie niskie wartości wskaźników insulinooporności w stosunku do chorych z otyłością centralną (163).
Jednakże w części prac nie wykazano jednoznacznej korelacji pomiędzy stężeniami HMW adiponektyny a insulinoopornością (164). Wyniki tych badań wskazują, że redukcja masy ciała i związany z nią wzrost insulinowrażliwości u osób stosujących niskokaloryczną dietę redukcyjną nie skutkował wzrostem stężeń adiponektyny całkowitej i HMW adiponektyny w surowicy krwi (165, 166). Z drugiej jednak strony, u osób po 60. r.ż. wykazano, że redukcja masy ciała uzyskana w wyniku włączenia diety niskokalorycznej oraz zwiększenia wysiłku fizycznego związana była z poprawą nie tylko insulinowrażliwości, ale także spowodowała wzrost wskaźnika HMW adiponektyna/adiponektyna całkowita oraz zwiększenie ekspresji receptorów dla adiponektyny w mięśniach (167). Wadą przytoczonych powyżej badań jest fakt, że były to obserwacje krótkotrwałe, trwające 10-12 tygodni, a zatem uzyskaną poprawę wskaźników metabolicznych trudno jest uznać za wyniki stabilne. Kolejnym ograniczeniem jest brak możliwości oceny utrzymania się poprawy parametrów (zarówno antropometrycznych, jak i biochemicznych) w dłuższym okresie obserwacji.
W piśmiennictwie istnieją doniesienia opierające się na badaniach oceniających wydzielanie adiponektyny wysokocząsteczkowej u osób otyłych (zarówno kobiet, jak i mężczyzn) w stosunku do osób szczupłych. Wykazano obniżone stężenia adiponektyny u osób otyłych (przy czym u kobiet w stosunku do mężczyzn zanotowano wyższe stężenia adiponektyny całkowitej i jej izoform zarówno u osób otyłych, jak i szczupłych). W pracy tej autorzy wykazali, że niższe poziomy adiponektyny i jej izoform związane były w większym stopniu z otyłością centralną niż z insulinoopornością (164).
W badaniach własnych stwierdzono, że obniżenie poziomu adiponektyny wysokocząsteczkowej u kobiet z nadwagą i otyłością (BMI ≥ 25 kg/m2) wydaje się być związane silniej ze wskaźnikami insulinooporności niż otyłości typu centralnego. Wskazuje na to silna ujemna korelacja pomiędzy HMW adiponektyną a wskaźnikiem HOMA-IR oraz stężeniem insuliny na czczo.
Dostępne wyniki badań wykonanych przez licznych autorów wskazują na potencjalny ochronny wpływ adiponektyny i jej wysokocząsteczkowej izoformy na wystąpienie chorób układu sercowo-naczyniowego (31, 68, 129, 168, 169). Poza kilkoma opublikowanymi w ostatnim roku pracami, a przede wszystkim publikacją Dekert i wsp. (36), w której wykazano, że wysokie stężenia adiponektyny mogą być predyktorem wzrostu ryzyka zgonu u osób z niewydolnością krążenia, większość danych wskazuje na zmniejszenie częstości incydentów sercowo-naczyniowych u osób z wyższymi stężeniami HMW adiponektyny. Z kolei, w pracy Sattar i wsp. opublikowanej w 2008 r. wskazuje się na brak zależności pomiędzy częstością incydentów sercowo-naczyniowych a poziomami adiponektyny wysokocząsteczkowej (170). Natomiast w badaniu oceniającym stężenia desacylowanej greliny i HMW adiponektyny wykazano, że to desacylowana grelina, a nie HMW adiponektyna jest potencjalnym predyktorem wystąpienia choroby niedokrwiennej serca (171).
Rezystyna
Wyniki badań rezystyny wykazały istotny statystycznie wzrost stężenia rezystyny w surowicy pacjentek z BMI ≥ 25 kg/m2 w stosunku do kobiet z grupy kontrolnej. W przeprowadzonych analizach statystycznych wykazano dodatnie korelacje pomiędzy rezystyną a BMI, BIA, obwodem talii i bioder oraz leptyną.
Rezystyna wydaje się odgrywać istotną rolę w patogenezie insulinooporności, otyłości i cukrzycy typu 2. Badania eksperymentalne na modelu zwierzęcym wskazują na ścisłą zależność pomiędzy wzrostem poziomu rezystyny a nasileniem insulinooporności. Jednakże wyniki badań dotyczących tego zagadnienia u ludzi nie są już tak jednoznaczne (22, 153, 172). Wydaje się, że istotne znaczenie dla efektu biologicznego wywieranego przez rezystynę ma miejsce jej syntezy. U zwierząt są to, przede wszystkim adipocyty, natomiast u ludzi są to głównie makrofagi i jednojądrowe komórki immunologiczne znajdujące się w zrębie tkanki tłuszczowej (173, 174). Badania koncentrujące się nad miejscem syntezy rezystyny wykazały, że jest ona produkowana głównie w wisceralnej tkance tłuszczowej, a w znacznie mniejszej ilości w podskórnej tkance tłuszczowej (21).
Badania prowadzone u ludzi in vivo oraz in vitro wskazują, że makrofagi zlokalizowane w tkance tłuszczowej pod wpływem takich czynników, jak TNFα, IL6, białka ostrej fazy (CRP) lub lipopolisacharydy bakterii Gram ujemnych zwiększają syntezę rezystyny. Z drugiej strony wykazano także, że rezystyna stymuluje ekspresję IL6 i TNFα w adipocytach białej tkanki tłuszczowej (21, 47). Potwierdzeniem ścisłego związku pomiędzy rezystyną a układem immunologicznym i czynnikami prozapalnymi są badania, w których zanotowano podwyższone stężenia rezystyny u osób z sepsą lub wstrząsem septycznym (175). Jednocześnie u chorych z przewlekłym procesem zapalnym w przebiegu reumatoidalnego zapalenia stawów wysokie stężenia rezystyny stwierdzane były w płynie stawowym, jak również w surowicy w porównaniu do osób z grupy kontrolnej (176, 177).
Liczne opublikowane prace kliniczne i eksperymentalne nie dały jednoznacznej odpowiedzi na pytanie dotyczące związku rezystyny z insulinoopornością oraz nie wyjaśniły dotychczas mechanizmów je regulujących. Nie można zatem jednoznacznie stwierdzić, czy wzrost stężenia rezystyny w otyłości lub cukrzycy typu 2 jest bezpośrednio związany z insulinoopornością.
Przeprowadzone badania własne wykazały najwyższe wartości rezystyny u kobiet z otyłością olbrzymią z towarzyszącym zespołem metabolicznym. Badania korelacji pomiędzy poziomami insuliny, glukozy czy wartościami HOMA-IR a poziomami rezystyny wykazywały dodatnie korelacje, jednakże w grupie pacjentek z BMI ≥ 25 kg/m2 nie były one znamienne statystycznie. Uzyskane wyniki badań własnych wskazują, że rezystyna może odgrywać istotną rolę w rozwoju insulinooporności, jednakże działanie jej nie wydaje się być bezpośrednie.
Grelina (Aktywna forma greliny – Ghrelin active)
Uzyskane wyniki badań własnych wykazały obniżenie poziomu aktywnej greliny u kobiet z BMI ≥ 25 kg/m2 w stosunku do pacjentek z grupy kontrolnej. Stwierdzono, że u pacjentek z nadwagą i otyłością poziomy aktywnej greliny korelowały ujemnie ze wskaźnikiem talia-biodra (WHR), obwodem talii, insuliną i HOMA-IR, a więc wykładnikami otyłości trzewnej i insulinooporności.
Analiza stężeń aktywnej greliny w poszczególnych badanych podgrupach kobiet z otyłością i nadwagą nie wykazała istotnych statystycznie różnic pomiędzy tymi grupami. Tylko u pacjentek z otyłością olbrzymią zanotowano znamiennie statystycznie niższe poziomy aktywnej greliny w porównaniu do wyników kobiet szczupłych z grupy kontrolnej. Jednakże po podzieleniu grupy kobiet z BMI ≥ 40 kg/m2 na 2 podgrupy w zależności od obecności zespołu metabolicznego różnice w zakresie stężeń aktywnej greliny nie wykazywały już znamienności statystycznej zarówno wewnątrz grupy otyłości olbrzymiej, jak i w porównaniu obu podgrup do grupy kontrolnej.
Grelina należy do peptydów oreksygenicznych i lipogenicznych, których poziom rośnie w czasie głodzenia, a po przyjęciu posiłku poziom greliny całkowitej ulega bardzo szybkiemu spadkowi (32, 71, 97, 178, 179). Peptyd ten w krwi obwodowej występuje w 2 głównych formach: w postaci acylowanej (zawierającą grupę oktanylową przy N terminalnym końcu) i w postaci desacylowanej, która stanowi ponad 80% greliny całkowitej. Formą aktywną greliny (grelin active) jest postać acylowana, która wywiera nieco inne działania metaboliczne niż grelina nieacylowana (97, 178, 179).
Wiadomo jest, że grelina u zwierząt i ludzi zwiększa łaknienie, stymuluje wydzielanie hormonu wzrostu i opróżnianie żołądka. U ludzi z prawidłowym BMI poziom greliny acylowanej i nieacylowanej ulega wzrostowi pod wpływem głodzenia i obniża się po przyjęciu posiłku (180). Z kolei, osoby otyłe charakteryzują się niższymi wyjściowymi poziomami greliny na czczo w stosunku do osób szczupłych (181). Ponadto wydaje się, że oznaczanie desacylowanej formy greliny może być użytecznym markerem miażdżycy u starszych pacjentów z nadciśnieniem tętniczym (171).
Korelacje greliny (formy aktywnej i nieaktywnej) ze wskaźnikami gospodarki węglowodanowej i wskaźnikami insulinooporności również nie są jednoznacznie określone w dostępnym piśmiennictwie. Stwierdzono, że wydzielanie greliny ulega zmniejszeniu w sytuacjach dodatniego bilansu energetycznego ustroju. Badania kliniczne wskazują na możliwość istnienia ujemnej korelacji pomiędzy greliną a BMI, zawartością tkanki tłuszczowej w organizmie, stężeniem glukozy, insuliny czy HOMA-IR (32, 71, 97, 178, 179). Wyniki badań własnych były zbieżne z przedstawionymi powyżej danymi. Poza tym badania przeprowadzone u zdrowych szczupłych ochotników nie wykazały wpływu greliny active na wskaźniki insulinooporności indukowanej krótkotrwałym podawaniem deksametazonu (182).
Badania opublikowane przez Żwirską-Korczałę i wsp. w 2007 r. (180) u kobiet otyłych z towarzyszącym zespołem metabolicznym i u pacjentek z otyłością olbrzymią nie wykazały pomiędzy tymi dwoma grupami istotnych statystycznie różnic w poziomach aktywnej greliny. Dodatkowo, u kobiet z otyłością olbrzymią nie zanotowano charakterystycznego poposiłkowego spadku poziomu aktywnej greliny. Autorzy tej pracy sugerują, że w tej szczególnej grupie pacjentek może dochodzić do swego rodzaju hipergrelinemii.
Wisfatyna
W uzyskanych wynikach badań własnych wykazano dodatnią korelację wisfatyny z BMI, BIA, obwodem talii i bioder oraz leptyną, a ujemną korelację z triglicerydami.
Ekspresja wisfatyny wykazana została, między innymi w białej tkance tłuszczowej, jak również w monocytach krwi obwodowej, czy też w makrofagach obecnych w podścielisku tkanki tłuszczowej. W części prac podkreślany jest fakt, że głównym miejscem syntezy i ekspresji wisfatyny jest wisceralna tkanka tłuszczowa i znajdujące się w jej zrębie komórki immunologicznie kompetentne. Postuluje się, że wisfatyna wykazuje też interakcje z innymi adipokinami, takimi jak rezystyna lub leptyna (49, 50).
Badania in vitro prowadzone na adipocytach linii komórkowej 3T3-L1 wskazują, że hipoksja powoduje zwiększoną ekspresję mRNA dla wisfatyny (183). Wydaje się, że wzrost poziomu wisfatyny w otyłości może być mechanizmem kompensacyjnym hipoksji tkanki tłuszczowej w przebiegu otyłości. Ponadto cytokiny prozapalne, takie jak IL6 lub TNFα zwiększają ekspresję wisfatyny w monocytach. Jednakże w adipocytach linii komórkowej 3T3-L1 poddanym dłuższemu działaniu lipopolisacharydów lub IL6 wykazano zjawisko „down regulation” dla wisfatyny (184, 185, 186).
Wzajemne relacje pomiędzy wisfatyną a zaburzeniami gospodarki węglowodanowej tkanką tłuszczową pozostają przedmiotem wielu badań.
W cukrzycy typu 2 stwierdzono podwyższone poziomy wisfatyny, jak również wykazano, że hiperglikemia może powodować wzrost poziomu tego peptydu w krwi obwodowej (49, 187).
W opublikowanych badaniach dotyczących korelacji wisfatyny z BMI istnieją dość duże rozbieżności (49, 51). W badaniach własnych potwierdzono dodatnią korelację poziomu wisfatyny z BMI.
W analizowanym piśmiennictwie dużo uwagi poświęcone zostało potencjalnemu insulinomimetycznemu działaniu wisfatyny. Podanie myszom tej adipokiny powoduje obniżenie poziomu glukozy w surowicy, a w przypadku myszy z obecną mutacją genu dla wisfatyny zanotowano wyższe wartości glikemii. Przypuszczalnie wisfatyna wywiera efekt addycyjny w stosunku do insuliny podczas łączenia się obu białek z receptorem insulinowym (49, 50, 51).
Prawdopodobnym jest również fakt, że podwyższone stężenia wisfatyny u osób z otyłością mogą być odzwierciedleniem dysfunkcji komórek tkanki tłuszczowej spowodowanej przewlekłym procesem zapalnym i podwyższonym poziomem cytokin, które w mechanizmie parakrynnym mogą stymulować produkcję wisfatyny.
Dostępne wyniki badania dotyczące zależności pomiędzy poziomem wisfatyny a profilem lipidowym pozostają rozbieżne. W części opublikowanych prac wykazano ujemną korelację stężeń wisfatyny i triglicerydów, co również wykazano w badaniach własnych, natomiast w niektórych pracach korelacja ta była dodatnia. U osób szczupłych z obecnością polimorfizmu genu wisfatyny SNP 1535 T/C z genotypem TT stwierdzano niższe poziomy wisfatyny w stosunku do osób z genotypem CC lub TC (52). Dane z części badań wskazują też na dodatnią korelację pomiędzy stężeniem wisfatyny w surowicy a HDL-cholesterolem, jednakże ze względu na istniejące rozbieżności w publikacjach obserwacje te wymagają dalszych szczegółowych analiz.
Otyłość olbrzymia (BMI ≥ 40 kg/m2)
W badanej grupie kobiet wykazano w stosunku do grupy kontrolnej podwyższone wartości wskaźników antropometryczych uzyskanych w badaniu przedmiotowym (skurczowe i rozkurczowe ciśnienie tętnicze krwi, BMI, BIA, obwód talii i bioder oraz WHR). Zanotowano charakterystyczne dla otyłości profile lipidowe pod postacią podwyższonych stężeń cholesterolu całkowitego, triglicerydów, LDL-cholesterolu i obniżonego poziomuHDL-cholesterolu.
U kobiet z otyłością olbrzymią, podobnie jak w całej analizowanej grupie pacjentek z BMI ≥ 25 kg/m2, wykazano podwyższone w stosunku do grupy kontrolnej wartości wskaźników gospodarki węglowodanowej (glukoza, insulina) oraz insulinooporności (HOMA-IR), natomiast w przypadku adipokin zanotowano znamiennie wyższe wartości leptyny, rezystyny, wisfatyny, indeksu wolnej leptyny, a obniżone wartości aktywnej greliny – adiponektyny i receptora leptynowego.
Adiponektyna i adiponektyna wysokocząsteczkowa (HMW – adiponektyna)
Analiza korelacji poszczególnych adipokin ze wskaźnikami antropometrycznymi i lipidowymi wykazała dodatnią korelację adiponektyny ze stężeniami HDL-cholesterolu będącą potwierdzeniem jej korzystnego wpływu na profil lipidowy oraz jej przeciwmiażdżycowego działania. Adiponektyna wykazywała ujemne korelacje ze skurczowym i rozkurczowym ciśnieniem tętniczym, WHR, HOMA-IR, cholesterolem całkowitym, LDL-cholesterolem i triglicerydami. Wyniki uzyskane u kobiet z otyłością olbrzymią były zbieżne z wynikami uzyskanymi w grupie kobiet z BMI ≥ 25 kg/m2 i potwierdzają korzystny wpływ adiponektyny na procesy metaboliczne.
Przeprowadzone badania wykazały także znamienne statystycznie niższe poziomy adiponektyny wysokocząsteczkowej w surowicy u pacjentek z otyłością olbrzymią w stosunku do grupy kobiet z prawidłową masą ciała z grupy kontrolnej. Wartości HMW adiponektyny u kobiet z BMI ≥ 40 kg/m2 były też znamiennie statystycznie niższe niż u kobiet z nadwagą i otyłością z zespołem metabolicznym i bez zespołu metabolicznego. Uzyskane wyniki dotyczące HMW adiponektyny były zbieżne z wynikami dotyczącymi adiponektyny całkowitej.
Opublikowane dotychczas wyniki badań eksperymentalnych i klinicznych dowodzą, że u osób z otyłością olbrzymią dochodzi do znacznej redukcji stężeń adiponektyny w surowicy i wartości te są silnej obniżone u mężczyzn niż u kobiet (188, 189). Zmniejszenie masy ciała u pacjentów z BMI ≥ 40 kg/m2 uzyskane w wyniku operacyjnego leczenia otyłości powoduje poprawę wskaźników insulinowrażliwości oraz wzrost poziomu adiponektyny wysokocząsteczkowej (190, 191). Dane dotyczące szybkości zmian stężeń HMW adiponektyny w trakcie redukcji masy ciała są niejednoznaczne. Wyniki większości prac wskazują, że wzrost poziomu wysokocząsteczkowej adiponektyny stwierdzany jest dopiero po ok. 3 miesiącach od zabiegu operacyjnego, a w obserwacjach długoterminowych (ok. 1 rok) wartości te wykazują tendencję do systematycznego wzrostu, co związane jest ze stopniową redukcją masy ciała (191, 192). Jednakże dane z niektórych badań wskazują, że wzrost poziomu tej adipokiny dotyczy, przede wszystkim adiponektyny całkowitej, adiponektyny średniocząsteczkowej lub wskaźnika HMW adiponektyna/adiponektyna całkowita (193). Długoterminowe obserwacje wykazują też, że przedoperacyjne wartości HMW adiponektyny mogą być też potencjalnym predyktorem ubytku masy ciała u chorych z otyłością olbrzymią przygotowywanych do leczenia operacyjnego otyłości (192).
Przeprowadzone badania własne u kobiet z otyłością olbrzymią potwierdzają korzystne korelacje pomiędzy adiponektyną wysokocząsteczkową a czynnikami wpływającymi na rozwój chorób układu sercowo-naczyniowego. Odnotowano bowiem ujemne korelacje ze skurczowym i rozkurczowym ciśnieniemm tętniczym, triglicerydami i LDL-cholesterolem, a dodatnią korelację z HDL-cholesterolem. HMW adiponektyna wykazywała także ujemne korelacje ze wskaźnikami insulinooporności (HOMA-IR i insulina). Uzyskane wyniki są zbieżne z danymi stwierdzanymi w piśmiennictwie i potwierdzają korzystne działanie metaboliczne adiponektyny wysokocząsteczkowej. Jak już wspomniano wcześniej, nie znany jest dokładny mechanizm korzystnego działania HMW adiponektyny powodujący obniżenie insulinooporności i ryzyka wystąpienia chorób układu sercowo-naczyniowego. Zatem problematyka ta wymaga dalszych badań.
Rezystyna
Rezystyna, oprócz podwyższonych stężeń w surowicy w stosunku do grupy kontrolnej, korelowała dodatnio z wartościami HOMA-IR, co może potwierdzać rolę w patogenezie insulinooporności. Natomiast porównanie wartości HOMA-IR we wszystkich analizowanych grupach pacjentek z nadwagą i otyłością wykazało, że najwyższe wartości HOMA-IR w badanym materiale stwierdzono w grupie kobiet z BMI 25-39,9 kg/m2 z towarzyszącym zespołem metabolicznym, a nie u kobiet z otyłością olbrzymią. Wynik ten można tłumaczyć częstością występowania zespołu metabolicznego w grupie kobiet z BMI ≥ 40 kg/m2, który rozpoznano jedynie u 73% pacjentek z otyłością olbrzymią. U pozostałych 27% pacjentek z otyłością olbrzymią nie występował zespół metaboliczny, w związku z tym pacjentki te miały znamiennie niższe wartości HOMA-IR.
Leptyna
Leptyna uważana jest za czynnik mający istotne znaczenie w patogenezie chorób układu sercowo-naczyniowego, cukrzycy i nadciśnienia tętniczego (67, 172, 194, 195). Większość opublikowanych badań wskazuje na to, że podwyższone wartości leptyny w surowicy krwi związane są ze zwiększonym ryzykiem udarów mózgu, wystąpieniem zawałów serca, insulinoopornością i cukrzycą typu 2. Poza tym podwyższone wartości indeksu wolnej leptyny korelują dodatnio z wyższymi wartościami ciśnienia tętniczego u osób bez otyłości i hiperglikemii (196).
Wyniki badań własnych u kobiet z otyłością olbrzymią wykazują odwrotne zależności pomiędzy wartościami ciśnienia tętniczego a poziomem leptyny w surowicy krwi (ujemna korelacja leptyna vs skurczowe i rozkurczowe ciśnienie tętnicze). Te zaskakujące wyniki mogą być potencjalnie związane z korzystnym wpływem leptyny na układ krążenia. Badania kliniczne wykazały bowiem, że leptyna może mieć działanie naczyniorozkurczające na tętnice wieńcowe u osób z chorobą niedokrwienną serca, jak również może powodować rozkurcz naczyń tętniczych przez stymulację produkcji NO w endotelium (150, 151, 197). Oprócz tego, podanie egzogennej leptyny nie wywołuje wzrostu ciśnienia tętniczego, pomimo udowodnionego pobudzającego wpływu na współczulny układ nerwowy, gdyż zwiększenie syntezy NO przez śródbłonek pod wpływem leptyny powoduje rozkurcz mięśniówki naczyniowej i zapobiega wzrostowi ciśnienia tętniczego (172, 194). Jednakże w badaniach klinicznych istnieją rozbieżności co do zależności pomiędzy podwyższonym stężeniem leptyny a ryzykiem chorób układu sercowo-naczyniowego. Romero-Corral i wsp. stwierdzili, że leptyna jest niezależnym czynnikiem chorób układu sercowo-naczyniowego mającym istotniejsze znaczenie niż białko C reaktywne (CRP), pomimo tego, że obydwa te markery były podwyższone w grupie osób z chorobami układu krążenia (198). Natomiast u osób z granicznymi wartościami ciśnienia tętniczego adiponektyna, a nie leptyna lub hsCRP, wydaje się być silniej skorelowana z ciśnieniem tętniczym krwi (199).
Wyniki badań własnych wskazują także na dodatnią korelację pomiędzy leptyną a HDL-cholesterolem i ujemną korelację z triglicerydami u pacjentek z otyłością olbrzymią. Podobnych wyników nie stwierdzono w innych analizowanych grupach kobiet.
Część badaczy koncentrujących swoje zainteresowania na problematyce otyłości, zaburzeń lipidowych i zaburzeń wydzielania adipokin stwierdza odwrotne zależności od uzyskanych i przedstawionych powyżej. Dane z tych prac pokazują, iż leptyna koreluje dodatnio z triglicerydami i ujemnie z HDL-cholesterolem. Badania uprzednio wykonywane w naszym ośrodku z udziałem osób powyżej 100-roku życia oraz osób z nadwagą i otyłością wykazały analogiczne wyniki (40). Podobnie w obecnym badaniu analiza wyników całej badanej grupy kobiet z BMI ≥ 25 kg/m2 wykazała ujemną korelację pomiędzy stężeniami leptyny i HDL-cholesterolu. Prace kliniczne, w których objęto obserwacją pacjentów leczonych z powodu otyłości wskazują, że redukcja masy ciała przy pomocy diety, leczenia farmakologicznego lub chirurgicznego skutkuje najczęściej obniżeniem poziomu leptyny w krwi obwodowej, czemu towarzyszy też poprawa parametrów profilu lipidowego.
Uzyskane paradoksalnie odwrotne korelacje pomiędzy leptyną a HDL-cholesterolem i triglicerydami w grupie pacjentek z otyłością olbrzymią mogą być związane ze zróżnicowanym oddziaływaniem leptyny w warunkach fizjologii i w stanach skrajnie patologicznych, jakim w szczególności jest otyłość olbrzymia. Badania Adami i wsp. wykazały, że leptyna może korelować dodatnio z HDL-cholesterolem (200). Natomiast u osób z otyłością olbrzymią może dochodzić do zmian wartości korelacji pomiędzy leptyną a parametrami lipidowymi w porównaniu do pacjentów z mniej nasiloną klinicznie otyłością (201).
Leptyna, adipokina syntetyzowana, przede wszystkim przez adipocyty podskórnej tkanki tłuszczowej przesyła do ośrodkowego układu nerwowego informacje dotyczące stanu odżywienia ustroju, ilości zgromadzonej tkanki tłuszczowej i pośrednio ilości zmagazynowanej energii. Fizjologicznie leptyna działa anoreksygenicznie i zwiększa wydatkowanie energii w mechanizmie, między innymi stymulacji receptorów β3 zlokalizowanych na adipocytach. Można uznać, iż leptyna jest białkiem przeciwdziałającym otyłości. Wydaje się jednak, że potencjalnie korzystna rola leptyny w otyłości polega na jej działaniu ochronnym poprzez wpływ na modulację akumulacji wolnych kwasów tłuszczowych w adipocytach, a w konsekwencji zabezpieczeniu innych tkanek i narządów przed toksycznym działaniem nadmiaru wolnych kwasów tłuszczowych (61, 68, 146). Ochrona przed opisanym powyżej zjawiskiem lipotoksyczności jest szczególnie istotna w przypadku komórek β wysp trzustkowych, kardiomiocytów i komórek mięśniowych gdyż lipotoksycznośc powoduje ich dysfunkcję, co, m.in. prowadzi do rozwoju insulinooporności. Innym antylipotoksycznym działaniem leptyny jest także stymulacja oksydacji wolnych kwasów tłuszczowych. Leptyna wykazuje również działanie lipolitycznie przez stymulacje drogi JAK/STAT3 i kinazy białkowej zależnej od AMP (AMPK) (202, 203, 204).
Udowodniono, że wraz z rozwojem otyłości dochodzi do zjawiska oporności na leptynę. Postuluje się, że przewlekle utrzymujące się podwyższone poziomy leptyny stwierdzane w otyłości powodują zaburzenie funkcji receptorów leptynowych w ośrodkowym układzie nerwowym, co skutkuje powstaniem leptynooporności. Z kolei leptynoooporność powoduje zaburzenie prawidłowej homeostazy energetycznej ustroju, „przesuwając” ją w kierunku akumulacji energii, a więc do gromadzenia tkanki tłuszczowej i nasilenia stopnia otyłości (138, 204).
Istotny wpływ na efekt metaboliczny leptyny ma stopień jej związania z rozpuszczalnym receptorem leptynowym, czego odzwierciedleniem jest indeks wolnej leptyny. U osób szczupłych większość leptyny znajdującej się w krążeniu obwodowym związana jest z białkami, przede wszystkim z rozpuszczalnym receptorem leptynowym, natomiast u osób otyłych większość krążącej leptyny jest w postaci niezwiązanej, co spowodowane jest obniżeniem stężenia rozpuszczalnego receptora leptynowego w surowicy u tych osób (205, 206, 207).
W obecnym badaniu najwyższe wartości leptyny, a jednoczęśnie najniższe stężęnia rozpuszczalnego receptora leptynowego zanotowano w grupie kobiet z otyłością olbrzymią. Jest więc możliwe, że u kobiet z BMI ≥ 40 kg/m2 wzrost poziomu leptyny całkowitej oraz leptyny niezwiązanej z rozpuszczalnym receptorem (indeks wolnej leptyny) prowadzi do przełamania leptynooporności i korzystnego metabolicznego działania tego peptydu.
Przedstawione i omówione powyżej wyniki wymagać będą w przyszłości dalszych badań w większych liczebnie grupach pacjentek.
Wisfatyna
U kobiet z otyłością olbrzymią stężenia wisfatyny były znamiennie wyższe w stosunku nie tylko do grupy kontrolnej, ale także w porównaniu do pozostałych analizowanych grup pacjentek z nadwagą i otyłością.
W dostępnym piśmiennictwie brak jest jednoznacznych danych dotyczących związku pomiędzy poziomem wisfatyny w otyłości olbrzymiej a BMI (49). W części prac u osób z otyłością olbrzymią stwierdzono znacząco podwyższone poziomy wisfatyny, które ulegały obniżeniu po operacji bariatrycznej (opaskowaniu żołądka) i redukcji masy ciała (208), natomiast w innych pracach wykazano pooperacyjny wzrost poziomu wisfatyny (209, 210, 211). Wyniki badań własnych nie wykazały znamiennych statystycznie korelacji pomiędzy wisfatyną a BMI u pacjentek z otyłością olbrzymią.
W obecnym badaniu wisfatyna wykazywała w grupie kobiet z BMI ≥ 40 kg/m2 ujemną korelację z triglicerydami i dodatnią z HDL-cholesterolem.
Wyniki badań innych autorów dotyczące korelacji poziomów wisfatyny ze wskaźnikami gospodarki lipidowej nie są jak dotychczas jednoznaczne. We Wstępie i poprzednich podrozdziałach dyskusji przedstawiono obecne poglądy na potencjalne relacje pomiędzy wisfatyną a triglicerydami, w których stwierdzane są duże rozbieżności. Dodatnie korelacje pomiędzy wisfatyną a HDL-cholesterolem są przedmiotem kontrowersji, jednakże w części prac uzyskano wyniki wskazujące na potencjalny korzystny wpływ wisfatyny na profil lipidowy (212, 213, 214). Wzrost poziomu HDL-cholesterolu w stosunku do podwyższania się poziomów wisfatyny może wiązać się ze stymulującym działaniem wisfatyny na wewnątrzkomórkową syntezę NAD (dwuneukleotydu adenino-nikotynowego), który związany jest z metabolizmem HDL-cholesterolu. Pobudzający wpływ wisfatyny na syntezę wewnątrzkomórkowego NAD jest korzystny dla zachowania prawidłowej funkcji komórek β wysp trzustkowych. Ponadto istnieją doniesienia, że insulinomimetyczne działanie wisfatyny prawdopodobnie związane jest ze stymulacją syntezy NAD (215, 216, 217). Dodatkowo, badania genetyczne wykazały również potencjalny wpływ polimorfizmów genu wisfatyny na stężenia HDL-cholesterolu. Stwierdzono bowiem, że osoby z polimorfizmem genu wisfatyny: 948 G/T w przypadku obecności allelu T i 1535 T/C w przypadku genotypu TT wykazywały znamiennie wyższe poziomy HDL-cholesterolu (52, 53).
Wyniki badań własnych wskazują na dodatnią korelację pomiędzy poziomami wisfatyny w surowicy a ilością tkanki tłuszczowej, natomiast wzrost stężęnia wisfatyny w krwi obwodowej wydaje się wpływać korzystnie na profil lipidowy osób z nadwagą i otyłością, co najbardziej zaznacza się u osób z otyłością olbrzymią.
Wyniki badań uzyskane u pacjentek z BMI ≥ 40 kg/m2 po wyodrębnieniu podgrup pacjentek z towarzyszącym zespołem metabolicznym i bez zespołu metabolicznego
Badania populacyjne wskazują, że do 30% pacjentów z otyłością może mieć prawidłowy profil metaboliczny a pacjenci ci określani są jako metabolicznie zdrowi. Osoby te charakteryzują się wysoką insulinowrażliwością, mniejszym obwodem talii, prawidłowym profilem lipidowym i normotensją (218). Badania kliniczne wskazują, że osoby otyłe – metabolicznie zdrowe mają znacznie niższe ryzyko powikłań sercowo-naczyniowych niż otyli z zaburzeniami metabolicznymi (218, 219). Dokładne kryteria oceny tej populacji nie zostały jednoznacznie zdefiniowane (218).
W obecnym badaniu wśród kobiet z BMI ≥ 40 kg/m2 stwierdzono, że 27% badanych pacjentek nie spełniało kryteriów rozpoznania zespołu metabolicznego, a więc można było je zaliczyć do grupy otyłych – metabolicznie zdrowych. U żadnej z tych pacjentek nie rozpoznano cukrzycy ani nadciśnienia tętniczego. W grupie kobiet z otyłością olbrzymią bez zespołu metabolicznego stwierdzono znacznie niższe wartości skurczowego i rozkurczowego ciśnienia tętniczego oraz WHR w stosunku do kobiet z otyłością olbrzymią i zespołem metabolicznym. Również wskaźniki gospodarki lipidowej wykazywały wyższe wartości HDL-cholesterolu i niższe triglicerydów. Opisane zmiany w profilu lipidowym należy odnosić, przede wszystkim do definicji zespołu metabolicznego wg kryteriów IDF 2005, w której wartości stężeń HDL-cholesterolu i triglicerydów zaliczone zostały do kryteriów rozpoznania. Poza tym w zakresie LDL-cholesterolu i cholesterolu całkowitego nie zanotowano istotnych statystycznie różnic.
Pacjentki z BMI ≥ 40 kg/m2 bez zespołu metabolicznego charakteryzowały się znacznie lepszymi wskaźnikami gospodarki węglowodanowej (niższe wartości glukozy i insuliny) i insulinooporności (niższe wartości HOMA-IR) w porównaniu do kobiet z otyłością olbrzymią i zespołem metabolicznym.
Analizując profil peptydowy, stwierdzono, że kobiety z BMI ≥ 40 kg/m2 bez zespołu metabolicznego miały niższe poziomy rezystyny (różnica niezamienna statystycznie), jak również istotnie statystycznie wyższe poziomy wisfatyny i adiponektyny w odniesieniu do kobiet z BMI ≥ 40 kg/m2 i zespołem metabolicznym. Natomiast wartości adiponektyny u kobiet z otyłością olbrzymią – metabolicznie zdrowych nie różniły się istotnie od wartości zanotowanych u kobiet szczupłych z grupy kontrolnej.
Poziomy adiponektyny wysokocząsteczkowej wykazywały tendencję do wyższych wartości u kobiet z otyłością olbrzymią bez zespołu metabolicznego w porównaniu do pacjentek z otyłością olbrzymią skojarzoną z zespołem metabolicznym, ale różnice te nie były znamienne statystycznie. Podobnie jak w przypadku adiponektyny całkowitej stężenia HMW adiponektyny u kobiet z BMI ≥ 40 kg/m2 bez zespołu metabolicznego nie różniły się istotnie od wyników uzyskanych w grupie kontrolnej. Należy podkreślić fakt, że kobiety z analogiczną masą ciała i towarzyszącym zespołem metabolicznym miały wartości HMW adiponektyny w surowicy znamiennie niższe od kobiet z prawidłowym BMI z grupy kontrolnej.
Przedstawione wyniki badań własnych wskazują na istotne różnice występujące wewnątrz grupy kobiet z otyłością w zakresie wydzielanych adipokin w zależności od tego, czy podstawowa choroba, jaką jest otyłość, jest skojarzona z zespołem metabolicznym. Osoby otyłe metabolicznie zdrowe charakteryzują się znacznie lepszymi wynikami nie tylko w zakresie WHR czy HOMA-IR, ale również posiadają korzystniejszy profil wydzielanych adipokin.
Badania innych autorów potwierdzają, że u osób otyłych – metabolicznie zdrowych stężenia adiponektyny są wyższe niż u osób otyłych z zaburzeniami metabolicznymi. Istotną rolę patogenetyczną w opisywanych powyżej zmianach odgrywa insulinooporność, a w licznych pracach udowodniono, że stężenia adiponektyny w surowicy korelują ujemnie z HOMA-IR i poziomami insuliny (38, 153, 220). Również w przedstawionych wynikach badań własnych potwierdzono ujemną zależność pomiędzy adiponektyną a wskaźnikami insulinooporności.
Wykazanie tendencji do wyższych, aczkolwiek niezamiennych statystycznie, wartości rezystyny u kobiet z BMI ≥ 40 kg/m2 i zespołem metabolicznym potwierdza istotną rolę insulinooporności oraz przewlekłego procesu zapalnego w tkance tłuszczowej w rozwoju zaburzeń metabolicznych w otyłości olbrzymiej.
Jednoznaczna interpretacja podwyższonych stężeń wisfatyny u kobiet BMI ≥ 40 kg/m2 bez zespołu metabolicznego w odniesieniu do insulinooporności jest utrudniona, między innymi ze względu na niewielką liczbę pacjentek w tej grupie, jednakże biorąc pod uwagę potencjalne insulinomimetyczne działanie tej adipokiny nie można wykluczyć jej korzystnego wpływu na zapobieganie rozwoju insulinooporności u pacjentek z otyłością olbrzymią.
Otyłość jest chorobą metaboliczną, której towarzyszy przewlekły proces zapalny mający miejsce w tkance tłuszczowej. Aspekt zapalenia często jest niedoszacowany w patomechanizmie powikłań otyłości.
Jak już zostało to zaznaczone we wcześniejszych rozdziałach, adiponektyna jest adipokiną o wieloprofilowej aktywności. Badania eksperymentalne udowodniły przeciwzapalne działanie adiponektyny wywierane przez receptor AdipoR1 (34, 153). Szczególnie istotnym wydaje się być supresyjny wpływ adiponektyny na ekspresję TNFα zarówno w wątrobie, jak i w tkance tłuszczowej. Wiadome jest, że TNFα odgrywa istotną rolę w rozwoju insulinooporności (221, 222). W warunkach in vitro adiponektyna stymuluje produkcję IL10 i IL1Ra (antagonista receptora IL1) w hodowlach ludzkich monocytów oraz hamuje w komórkach immunologicznie kompetentnych kompleks NF-κB – odpowiedzialny za syntezę cytokin prozapalnych (223, 224, 225). Wydaje się, że adiponektyna ma modulujący wpływ na proces zapalny towarzyszący nadmiernemu gromadzeniu się tkanki tłuszczowej. Nie należy pomijać jednak wyników niektórych doświadczeń, w których w warunkach eksperymentalnie wywołanego zapalenia adiponektyna wydawała się być czynnikiem prozapalnym (226). Rozbieżności te wymagają dalszych badań, w tym szczegółowych badań klinicznych.
Udowodnione jest, że podwyższone poziomy adiponektyny wpływają także korzystnie na profil lipidowy przez nasilenie akumulacji lipidów w adipocytach i zapobieganie w ten sposób zjawisku lipotoksyczności (34).
Wysokocząsteczkowa adiponektyna (HMW adiponektyna) uznawana jest za najaktywniejszą biologicznie frakcję adiponektyny i w większości badań jej stężenie w surowicy koreluje ujemnie ze wskaźnikami insulinooporności. Wiadomo jest, że stosowanie leków z grupy tiazolidinedionów powoduje nie tylko wzrost insulinowrażliwości, ale także skutkuje istotnym podwyższeniem stężenia HMW adiponektyny w surowicy (34, 57). Jednakże podanie leków z tej grupy myszom nie syntetyzującym adiponektyny nie powoduje poprawy parametrów metabolicznych oraz insulinowrażliwości (227). Można zatem stwierdzić, że spadek insulinooporności koreluje ze wzrostem wysokocząsteczkowej adiponektyny, co potwierdzono także w obecnych badaniach własnych. Mechanizm działania leków z grupy tiazolidinedionów na syntezę adiponektyny całkowitej i adiponektyny wysokocząsteczkowej związany jest z aktywacją receptorów PPARγ. Wydaje się, że tiazolidinediony stymulują, przede wszystkim wzrost adiponektyny wysokocząsteczkowej, a w mniejszym stopniu adiponektyny całkowitej. Korzystnym metabolicznie działaniem jest to, że HMW adiponektyna zmniejsza produkcję endogennej glukozy, co powoduje obniżenie insulinooporności (34, 57, 68, 153).
Na podstawie wyników uzyskanych w obecnym badaniu można uznać, że zarówno adiponektyna wysokocząsteczkowa, jak i adiponektyna całkowita wykazują istotne zmiany w otyłości olbrzymiej w zależności od tego, czy choroba ta współistnieje z zespołem metabolicznym. Tak więc podział kliniczny pacjentów otyłych na otyłych metabolicznie zdrowych i z zaburzeniami metabolizmu ma istotne uzasadnienie kliniczne.
Rola wisfatyny w przebiegu procesu zapalnego, a w szczególności w zapaleniu toczącym się w tkance tłuszczowej, pozostaje dotychczas niewyjaśniona. Uważa się na podstawie badań in vitro, że jest to adipokina o działaniu prozapalnym stymulująca syntezę IL7, IL6, TNFα i IL1β w hodowlach ludzkich monocytów (49, 228). Dodatkowo, wisfatyna aktywuje limfocyty T i monocyty oraz nasila syntezę czynników chemotaktycznych. Jednakże stosowanie wysokich dawek wisfatyny stymuluje syntezę cytokin przeciwzapalnych, takich jak IL10 lub IL1Ra (228, 229).
W oparciu o porównanie wyników badań innych badaczy prowadzonych u otyłych pacjentów, u których w wyniku leczenia doprowadzono do redukcji masy ciała, wykazano, że obniżenie masy ciała prowadzi do spadku wartości CRP i LDL-cholesterolu, ale tylko u pacjentów otyłych z towarzyszącym zespołem metabolicznym, natomiast u osób otyłych – zdrowych metabolicznie nie zanotowano istotnych zmian w zakresie opisywanych powyżej parametrów, pomimo uzyskania redukcji masy ciała (230). Jest więc prawdopodobne, że u osób otyłych lecz zdrowych metabolicznie odczyn zapalny w tkance tłuszczowej jest znacznie mniej nasilony, co skutkuje korzystniejszym profilem lipidowym, lepszymi wskaźnikami insulinooporności oraz znacznie mniejszymi zaburzeniami wydzielania adipokin niż u otyłych z rozpoznanym zespołem metabolicznym. Potwierdzeniem tej teorii mogą być badania immunologiczne wykonane u pacjentów z otyłością olbrzymią z obecnym lub nie zespołem metabolicznym, które wykazały iż otyli z BMI ≥ 40 kg/m2 zdrowi metabolicznie mają znamiennie więcej limfocytów NK i limfocytów cytotoksycznych niż otyli z BMI ≥ 40 kg/m2 skojarzonym z zespołem metabolicznym, aczkolwiek obie opisane powyżej grupy charakteryzowały się gorszymi parametrami niż osoby szczupłe (231).
W innych badaniach eksperymentalnych wykonanych na otyłych szczurach rasy Zucker (tzw. szczurach fa/fa, posiadających mutację w genie kodującym receptor leptynowy) wykazano, że nadmierna ekspresja receptora dla angiotensyny 1 (AT-1R) w śródbłonku naczyń może odpowiadać za rozwój zaburzeń prowadzących do nasilenia miażdżycy i zespołu metabolicznego (232).
Przedstawione wyniki badań własnych oraz dane uzyskane z piśmiennictwa wskazują na istotną rolę odczynu zapalnego i odpowiedzi immunologicznej na rozwój insulinooporności i powikłań metabolicznych związanych z otyłością. W przypadku otyłości olbrzymiej można stwierdzić, że spośród analizowanych adipokin adiponektyna ma najistotniejsze działanie ochronne wobec powikłań związanych z insulinoopornością. Uzyskane wyniki badań dotyczące wisfatyny wskazują na jej potencjalny korzystny wpływ na profil lipidowy, a w konsekwencji na możliwe działanie przeciwmiażdżycowe. Wyodrębnienie u osób z otyłością olbrzymią pacjentów bez towarzyszącego zespołu metabolicznego może okazać się prostym i uzasadnionym postępowaniem klinicznym, gdyż pacjenci ci mają znacznie lepsze wyniki badań metabolicznych, a w konsekwencji mniejsze ryzyko powikłań sercowo-naczyniowych.
Nadwaga i otyłość (BMI 25-39,9 kg/m2) z towarzyszącym zespołem metabolicznym
Wyniki uzyskane u kobiet z BMI 25-39,9 kg/m2 i towarzyszącym zespołem metabolicznym w porównaniu do grupy kontrolnej wykazały podobne zmiany w zakresie wyników badań lipidowych oraz parametrów oceniających gospodarkę węglowodanową i insulinooporność, jak w przypadku otyłości olbrzymiej.
Również wszystkie analizowane adipokiny wykazały istotne statystycznie różnice w stosunku do grupy kontrolnej. Wartości stężeń w surowicy były znamiennie wyższe w przypadku leptyny, wisfatyny, indeksu wolnej leptyny, a adiponektyny całkowitej i adiponektyny wysokocząsteczkowej (HMW) istotnie niższe w odniesieniu do grupy kontrolnej. Ponadto adiponektyna korelowała dodatnio HDL-cholesterolem i ujemnie z WHR, obwodem talii, stężeniem insuliny, HOMA-IR i triglicerydami.
Uzyskane wyniki są zgodne z wynikami przedstawianymi w literaturze dotyczącymi ujemnych korelacji pomiędzy stężeniami w surowicy adiponektyny a wskaźnikami gospodarki węglowodanowej i insulinooporności.
Wykonana analiza uzyskanych wyników badań u kobiet z BMI 25-39,9 kg/m2 skojarzonym z zespołem metabolicznym wskazuje, że obniżenie stężenia adiponektyny w tej podgrupie w stosunku do grupy kontrolnej jest najprawdopodobniej związane z najwyższymi wartościami wskaźników gospodarki węglowodanowej i insulinooporności (insulina i glukoza na czczo oraz HOMA-IR) spośród wszystkich ocenianych pacjentek z nadwagą i otyłością.
Ujemna korelacja adiponektyny ze wskaźnikiem WHR oraz obwodem talii sugeruje, że obniżenie stężenia adiponektyny związane może być ze współistnieniem otyłości typu wisceralnego.
Badania przeprowadzone u kobiet leczonych z powodu otyłości wykazały obniżenie poziomu adiponektyny i wysokocząsteczkowej adiponektyny w surowicy u kobiet z otyłością typu centralnego (57, 68).
Uzyskane w badaniu własnym w tej podgrupie pacjentek podwyższone stężenia leptyny i rezystyny korelowały dodatnio z BMI i wyniki te są zgodne z większością publikowanych danych. Natomiast w przypadku wisfatyny nie wykazano korelacji związanych z parametrami gospodarki lipidowej, analogicznych do grupy kobiet z otyłością olbrzymią. Wisfatyna korelowała dodatnio z BMI, BIA, obwodem talii i bioder.
Podsumowując wyniki badań uzyskanych u kobiet z BMI 25-39,9 kg/m2 z towarzyszącym zespołem metabolicznym, można uznać, że są one zgodne z wynikami prezentowanymi w piśmiennictwie i wskazują na ścisły związek pomiędzy nasileniem insulinooporności a zespołem metabolicznym, jak również implikują modulujący wpływ insulinooporności na wydzielanie adipokin, a przede wszystkim na wydzielanie adiponektyny (14, 57, 68, 153).
Nadwaga i otyłość (BMI 25-39,9 kg/m2) bez towarzyszącego zespołu metabolicznego
Podobnie jak w przypadku omawianych poprzednio grup pacjentek, kobiety z BMI 25-39,9 kg/m2 bez zespołu metabolicznego miały wskaźniki antropometryczne (BMI, BIA, obwód talii i bioder, WHR) i skurczowe ciśnienie tętnicze krwi znamiennie statystycznie wyższe w porównaniu do grupy kontrolnej. Wyjątek stanowiło rozkurczowe ciśnienie tętnicze, którego wartości nie różniły się istotnie od grupy kontrolnej.
Wskaźniki gospodarki lipidowej, węglowodanowej i insulinooporności były zaburzone podobnie jak w poprzednich analizowanych grupach kobiet z otyłością w odniesieniu do grupy kontrolnej.
Analiza statystyczna uzyskanych stężeń adipokin w surowicy wykazała znamiennie statystycznie wyższe wartości leptyny, indeksu wolnej leptyny w stosunku do grupy kontrolnej oraz tendencję do wyższych wartości wisfatyny i rezystyny, jednakże różnice te nie wykazywały istotności statystycznej.
Natomiast w przypadku adiponektyny i adiponektyny wysokocząsteczkowej uzyskano wyniki nieróżniące się od wyników zanotowanych w grupie kontrolnej składającej się z młodych szczupłych kobiet.
Analizowana grupa kobiet spełniała podstawowe kryterium do rozpoznania nadwagi i otyłości, a więc BMI ≥ 25 kg/m2, jednakże pacjentki z tej podgrupy nie spełniały kryteriów rozpoznania zespołu metabolicznego, zatem możemy je określić jako metabolicznie zdrowe. Brak różnic w poziomach adiponektyny, adiponektyny wysokocząsteczkowej i rezystyny w surowicy pomiędzy grupą kontrolną a kobietami z nadwagą i otyłością (BMI 25-39,9 kg/m2) bez zespołu metabolicznego potwierdza fakt istnienia otyłości, w której zaburzenia metaboliczne są znacznie mniej nasilone, co może skutkować zmniejszonym ryzykiem powikłań sercowo-naczyniowych (218, 233).
Porównanie wyników uzyskanych w grupach kobiet z BMI 25-39,9 z towarzyszącym zespołem metabolicznym i bez zespołu metabolicznego
Porównując wyniki wskaźników antropometrycznych w wymienionych powyżej grupach kobiet, zanotowano wyższe wartości skurczowego i rozkurczowego ciśnienia tętniczego oraz podwyższone wskaźniki otyłości centralnej (obwód talii i WHR) u kobiet z otyłością skojarzoną z zespołem metabolicznym. Wymienionym zmianom stwierdzonym w badaniu przedmiotowym towarzyszyły podwyższone poziomy insuliny, glukozy i wskaźnika HOMA-IR w przepadku współwystępowania zespołu metabolicznego i otyłości. Podobnie niekorzystne zmiany dotyczyły profilu lipidowego, gdyż u kobiet otyłych z zespołem metabolicznym wartości HDL-cholesterolu były niższe, a cholesterolu całkowitego, LDL-cholesterolu i triglicerydów wyższe niż u kobiet z otyłością metabolicznie zdrowych.
Badania eksperymentalne i kliniczne wskazują, że tkanka tłuszczowa zlokalizowana w górnej połowie ciała (tkanka tłuszczowa brzuszna wisceralna i podskórna) wykazuje inne właściwości metaboliczne niż tkanka tłuszczowa zlokalizowana w okolicy pośladkowo-udowej (21, 68, 138). Podskórna tkanka tłuszczowa zlokalizowana w górnej połowie ciała charakteryzuje się znacznie mniejszą możliwością akumulacji triglicerydów oraz wolniejszym metabolizmem wolnych kwasów tłuszczowych. Natomiast tkankę tłuszczową wisceralną cechuje większa aktywność lipolityczna i zwiększona zdolność do uwalniania wolnych kwasów tłuszczowych w przebiegu hiperinsulinizmu i insulinooporności. Badania wskazują również, że adipocyty brzusznej tkanki tłuszczowej wykazują oporność na antylipolityczne działanie insuliny (21, 68, 138).
Obecnie w piśmiennictwie rozważana jest hipoteza, że wisceralna tkanka tłuszczowa jest miejscem „ektopowego magazynowania” nadmiaru tłuszczu podobnie jak ma to miejsce w przypadku magazynowania nadmiaru triglicerydów w miocytach lub w komórkach wątroby. Zjawisko to może być związane z dysfunkcją i zmniejszonymi możliwościami gromadzenia lipidów przez podskórną tkankę tłuszczową (14, 21, 57, 68, 138, 234).
Przeprowadzone badania własne, w których wykazano podwyższone parametry oceniające otyłość typu wisceralnego i towarzyszący im wzrost wskaźników insulinooporności u otyłych kobiet z rozpoznanym zespołem metabolicznym w stosunku do otyłych kobiet metabolicznie zdrowych, potwierdzają istotną rolę redystrybucji tkanki tłuszczowej w mechanizmie rozwoju powikłań metabolicznych otyłości.
Oceniając stężenia badanych adipokin, stwierdzono istotne statystycznie różnice w badanej populacji otyłych kobiet, w zależności od obecności lub braku zespołu metabolicznego. Kobiety, u których otyłość powikłana była współistnieniem zespołu metabolicznego, miały znamiennie niższe wartości wskaźnika wolnej leptyny w stosunku do otyłych kobiet bez rozpoznanego zespołu metabolicznego (natomiast różnice stężeń leptyny pomiędzy omawianymi grupami nie były znamienne statystycznie). Podobną sytuację zaobserwowano też we wcześniej analizowanej grupie pacjentek z BMI ≥ 40 kg/m2, gdzie wartości leptyny i wskaźnika wolnej leptyny u kobiet z otyłością olbrzymią bez towarzyszącego zespołu metabolicznego wykazywały tendencję do wartości wyższych (jednakże bez istotnych statystycznie różnic) niż u kobiet z BMI ≥ 40 kg/m2 z towarzyszącym zespołem metabolicznym.
W warunkach fizjologicznych leptyna hamuje wydzielanie insuliny (co potwierdzono, między innymi u myszy ob./ob. charakteryzujących się brakiem możliwości syntezy leptyny), natomiast insulina stymuluje wydzielanie leptyny (56, 67). Być może odnotowane tendencje do wyższych stężeń leptyny i indeksu wolnej leptyny u kobiet otyłych bez zespołu metabolicznego wpływały hamująco na wydzielanie insuliny i w ten sposób powodowały zmniejszenie insulinooporności u pacjentek otyłych metabolicznie zdrowych.
Analiza uzyskanych wyników stężeń adiponektyny w surowicy wykazała istotne statystycznie obniżenie jej wartości w podgrupie osób otyłych z zespołem metabolicznym w odniesieniu do kobiet otyłych bez zespołu metabolicznego (otyłych metabolicznie zdrowych).
Publikowane prace wskazują, że podstawową przyczyną obniżenia się wartości adiponektyny u kobiet z otyłością skojarzoną z zespołem metabolicznym (zarówno w porównaniu do grupy kontrolnej, jak i kobiet otyłych metabolicznie zdrowych) jest fakt współistnienia otyłości wisceralnej i insulinooporności (14, 34, 46, 153, 220).
Oprócz wymienionych w poprzednich rozdziałach wzajemnych zależności, ważnym pośrednim dowodem na istotny związek pomiędzy stężeniami adiponektyny a stopniem insulinowrażliwosci jest fakt stwierdzenia wyższych poziomów adiponektyny całkowitej u osób z genetycznie uwarunkowaną otyłością w przebiegu zespołu Willego-Pradera. W zespole tym wykazano wyższe poziomy adiponektyny całkowitej i HMW adiponektyny w porównaniu do osób z otyłością prostą. Ponadto u osób z zespołem Willego-Pradera stwierdzono także znamiennie niższe wskaźniki insulinooporności (163). Wyniki publikowanych prac wskazują na hamujący wpływ otyłości wisceralnej na syntezę adiponektyny w tkance tłuszczowej, jak również, że ekspresja adiponektyny w tkance tłuszczowej wisceralnej jest mniejsza niż w tkance podskórnej. Wiadomo również, że stosowanie agonistów receptorów PPARγ stymuluje syntezę adiponektyny w adipocytach oraz zwiększa insulinowrażliwość tkanek obwodowych (26, 57, 68, 235, 236).
Porównanie stężeń wysokocząsteczkowej adiponektyny w surowicy pomiędzy pacjentkami otyłymi z zespołem metabolicznym a otyłymi – metabolicznie zdrowymi wykazało tendencje do wyższych wartości HMW adiponektyny u kobiet otyłych metabolicznie zdrowych, jednakże różnice te nie były znamienne statystycznie.
Opublikowane dotychczas dane wskazują, że poszczególne frakcje adiponektyny zmniejszają reakcję zapalną przebiegającą w zrębie tkanki tłuszczowej, co przyczynia się do modulacji insulinooporności. Frakcje LMW (low molecular weight) i HMW adiponektyny wpływają na aktywność monocytów zlokalizowanych w zrębie tkanki tłuszczowej przez nasilenie ich apoptozy, aktywację AMPK (kinazy zależnej od AMP). Wpływ adiponektyny, w tym jej frakcji wysokocząsteczkowej na procesy zapalne i regulację odpowiedzi zapalnej, nie jest jednak do końca wyjaśniony (129, 146, 155, 237). Sugeruje się, że adiponektyna może stymulować aktywność cyklooksygenazy (COX2) typu 2 w kardiomiocytach, co prowadzi do wzrostu poziomu prostaglandyny E2 oraz obniżenia poziomu TNFα, co w konsekwencji powoduje zmniejszenie odczynu zapalnego w tkance tłuszczowej (238).
Poza zaburzoną syntezą adiponektyny i jej poszczególnych frakcji, a w szczególności frakcji wysokocząsteczkowej, należy zaznaczyć również fakt zaburzonej ekspresji receptorów adiponektyny w otyłości, co powoduje istotne konsekwencje kliniczne i metaboliczne wynikające z zaburzonej utylizacji glukozy i lipidów w wątrobie i mięśniach (24, 26).
Biorąc pod uwagę przedstawione powyżej fakty oraz udowodnione przeciwzapalne właściwości adiponektyny, wydaje się, że adipokina ta stanowić może istotny czynnik ochronny wobec chorób układu sercowo-naczyniowego, które występują u osób otyłych.
Dyskusja wyników badań genetycznych
Badania polimorfizmów genu adiponektyny
Przeprowadzone badania w grupie kobiet z BMI ≥ 25 kg/m2, analizowanej całościowo, nie wykazały istotnych statystycznie różnic w częstości występowania poszczególnych genotypów we wszystkich badanych polimorfizmach genu adiponektyny. Podobne wyniki uzyskano po przeprowadzeniu podziału osób badanych na poszczególne podgrupy kobiet z nadwagą i otyłością.
Natomiast badanie ilorazu szans (OR) w modelu dominującym uwidoczniło istotne statystycznie różnice w przypadku polimorfizmu 276 G/T genu adiponektyny u pacjentek z otyłością olbrzymią i w grupie chorych z nadwagą i otyłością skojarzoną z zespołem metabolicznym w stosunku do grupy kontrolnej kobiet szczupłych. Wyniki badania OR w tym modelu dla pozostałych polimorfizmów genu adiponektyny nie były znamienne statystycznie.
Dostępne w piśmiennictwie wyniki badań dotyczących wpływu polimorfizmów genu adiponektyny na jej stężenie w surowicy oraz związane z polimorfizmami ryzyko takich chorób, jak cukrzyca typu 2, insulinooporność, nadciśnienie tętnicze nie dały jednoznacznej odpowiedzi na pytanie o potencjalny wpływ czynników genetycznych na występowanie wymienionych powyżej chorób (31, 117, 130).
Polimorfizm pojedynczego nukleotydu 276 G/T genu adiponektyny wydaje się mieć znaczenie w zwiększeniu ryzyka chorób skojarzonych z niższymi stężeniami adiponektyny. Obecność allelu G zamiast allelu T w genotypie pacjentów powoduje hipoadiponektynemię, jak również wzrost ryzyka zachorowania na cukrzycę typu 2 (239, 240). Jednakże badania przeprowadzone na Tajwanie wśród osób powyżej 65. roku życia wykazały, że obecność allelu G polimorfizmu genu adiponektyny 276 G/T była związana z niższym ryzykiem występowania otyłości, zespołu metabolicznego i cukrzycy (241). Należy zaznaczyć fakt, ze nawet badania prowadzone w tych samych populacjach dają nierzadko rozbieżne wyniki (130).
Innym często opisywanym polimorfizmem genu adiponektyny skojarzonym ze zwiększonym ryzykiem chorób metabolicznych, insulinooporności oraz cukrzycy typu 2 jest polimorfizm 45 T/G, który może występować łącznie z polimorfizmem 276 G/T (242, 243, 244, 245). W badaniach własnych nie wykazano istotnych statystycznie zmian w zakresie OR SNP 45 T/G. Badania polskie opublikowane w 2009 r. wykazały, że polimorfizmy genu adiponektyny 11 391 G/A i 276 G/T były związane z wystąpieniem cukrzycy typu 2 (132).
Uzyskane wyniki SNP 276 G/T u kobiet z otyłością olbrzymią i w grupie kobiet z nadwagą i otyłością skojarzoną z zespołem metabolicznym wskazują, że niekorzystny genotyp GG występuje częściej w tych grupach w stosunku do grupy kontrolnej kobiet młodych z prawidłowym BMI. Istotne statystycznie zmiany ilorazu szans (OR) wskazują, że obecność allelu T w genotypie (homozygota TT lub heterozygota GT) wiąże się z mniejszym ryzykiem wystąpienia otyłości olbrzymiej lub otyłości skojarzonej z zespołem metabolicznym (ryzyko to jest mniejsze ok. 2,5 razy). W części prac sugeruje się, że poszczególne genotypy występujące w danym polimorfizmie mogą wpływać na stężenia adiponektyny w surowicy (239, 242, 245), jednakże w innych doniesieniach nie potwierdzono tego przypuszczenia (246, 247). Natomiast w przypadku porównania ilorazu szans pomiędzy grupami kobiet z nadwagą i otyłością z zespołem metabolicznym w stosunku do kobiet z nadwagą i otyłością bez zespołu metabolicznego nie uzyskano istotnych statystycznie wyników ilorazu szans w zakresie omawianego polimorfizmu.
W pozostałych analizowanych polimorfizmach genu adiponektyny nie stwierdzono istotnych statystycznie różnic w częstości występowania poszczególnych genotypów ani istotnych statystycznie różnic wyników badania ilorazu szans.
Dane w piśmiennictwie są w tym zakresie rozbieżne i często zależą od analizowanej populacji pacjentów. W populacji francuskiej obecność SNP 11 377 i 11 391 wiązała się z potencjalnym ryzykiem wystąpienia cukrzycy typu 2 i insulinoopornością (161, 244). Natomiast stwierdzenie SNP 276 G/T i 11 377 C/G może współistnieć ze zwiększonym ryzykiem otyłości olbrzymiej (248).
W analizowanym materiale własnym iloraz szans (OR) dla modelu dominującego polimorfizmu 11 377 C/G był bliski znamienności statystycznej i wskazywał na potencjalnie korzystny wpływ allelu G w otyłości olbrzymiej. Opublikowane dotychczas badania dotyczące tego polimorfizmu są rozbieżne ponieważ u pacjentów chorujących na cukrzycę w populacji francuskiej w SNP 11 377 C/G dominuje allel G, natomiast w populacji japońskiej i szwedzkiej u chorych z cukrzycą częściej stwierdzany jest allel C (161, 239, 249, 250).
Uzyskane wyniki badań własnych nie wykazały istotnych różnic w zakresie ilorazu szans oraz rozkładu genotypów pomiędzy grupami pacjentek z nadwagą i otyłością z towarzyszącym lub bez zespołu metabolicznego. Wyniki uzyskane w odniesieniu do grupy pacjentek młodych szczupłych są trudne do jednoznacznej interpretacji, gdyż porównywane grupy różnią się istotnie wiekiem i niemożliwe jest określenie, u której z kobiet z grupy kontrolnej wystąpi w przyszłości nadwaga lub otyłość.
Badania polimorfizmów genu rezystyny
W przeprowadzonych badaniach 3 polimorfizmów genu rezystyny nie stwierdzono istotnych zmian w częstości występowania poszczególnych genotypów dla każdego analizowanego polimorfizmu w stosunku do grupy kontrolnej. Wykazano natomiast znamienne statystycznie różnice ilorazu szans (OR) polimorfizmu genu rezystyny 420 C/G przy porównaniu pacjentek z nadwagą i otyłością skojarzoną z zespołem metabolicznym w stosunku do kobiet szczupłych z prawidłowym BMI. Badania własne wykazały, że genotyp GG lub CG może wiązać się ze zwiększonym ryzykiem wystąpienia otyłości i zespołu metabolicznego. Nie stwierdzono natomiast istotnych statystycznie zmian OR w zakresie analizowanych polimorfizmów pomiędzy pacjentkami z nadwagą i otyłością ze współistniejącym zespołem metabolicznym w stosunku do kobiet otyłych bez zespołu metabolicznego.
Dane z piśmiennictwa dotyczące znaczenia klinicznego polimorfizmów genu rezystyny są rozbieżne. Jednym z najczęściej analizowanych polimorfizmów tego genu jest RETN-420 C/G, którego obecność w populacji chińskiej może wiązać się ze wzrostem ryzyka wystąpienia choroby niedokrwiennej serca (wzrost ryzyka choroby wieńcowej dotyczy pacjentów z genotypem GG lub CG w stosunku do osób z genotypem CC) (42). W przeciwieństwie do wyników tej pracy badania autorów włoskich nie potwierdziły związku omawianego polimorfizmu z występowaniem zwiększonego ryzyka zachorowania na choroby układu sercowo-naczyniowego, zawał serca i niewydolność nerek (251). Natomiast badania wykonane w Japonii wykazały, że obecność polimorfizmu 420 C/G genu rezystyny korelowały z obniżonymi wartościami HDL-cholesterolu w surowicy i wzrostem białka C-reaktywnego, a więc markerów zwiększonego ryzyka chorób układu sercowo-naczyniowego (252).
Badania osób żyjących w południowych Chinach wykazały, że obecność allelu A w polimorfizmie genu rezystyny 62 G/A i allelu G w polimorfizmie 420 C/G może być czynnikiem wskazującym na ryzyko postępującego zwiększania się i stężenia glikemii w surowicy w czasie 5. letniej obserwacji (43). Natomiast badania polimorfizmu 62 G/A genu rezystyny wykazały w populacji niemieckiej jego związek z nadciśnieniem tętniczym, ale nie z cukrzycą typu 2 (136).
Wyniki obserwacji klinicznych dotyczących związków polimofizmów genu rezystyny a występowaniem otyłości pozostają także rozbieżne. W badaniach brazylijskich wykazano, że w przypadku 420 C/G kobiety homozygoty CC mają znacznie wyższe BMI niż nosicielki allelu G (253). Natomiast w populacji kanadyjskiej u mężczyzn z genotypem GG polimorfizmu 420 C/G stwierdzana jest mniejsza ilość wisceralnej tkanki tłuszczowej i mniej nasilony wyrzut insuliny w trakcie testu z doustnym obciążeniem glukozą (OGTT) (254). Autorzy tej publikacji nie potwierdzili podobnych wyników u kobiet, co może wskazywać na wpływ płci na parametry kliniczne i biochemiczne w przypadku występowania poszczególnych genotypów danego polimorfizmu genu rezystyny.
W badaniach opublikowanych w 2004 r. sugeruje się, że polimorfizmy 537 A/G i 420 C/G mogą wpływać na stężenie rezystyny w surowicy krwi, ale nie wykazano ich związku z ryzykiem zachorowania na cukrzycę typu 2 (255). Natomiast badania belgijskie nie wykazały związku polimorfizmów 420 C/G i IVS + 181 G/A genu rezystyny z wystąpieniem otyłości (256).
W analizowanym piśmiennictwie wpływ polimorfizmów genu rezystyny na ryzyko wystąpienia otyłości, nadciśnienia tętniczego czy zmian w lipidogramie lub wskaźnikach gospodarki węglowodanowej nie jest jednoznacznie wyjaśniony.
Analiza regresji logistycznej
Nie wykazano istotnych statystycznie asocjacji badanych polimorfizmów w przypadku analizy kobiet z otyłością i nadwagą z zespołem metabolicznym w stosunku do kobiet z otyłością i nadwagą bez zespołu metabolicznego.
Wyniki przeprowadzonej analizy regresji logistycznej przy założeniu jako zmiennej zależnej nadwagi i otyłości z zespołem metabolicznym wykazały asocjację z otyłością i zespołem metabolicznym 3 badanych polimorfizmów (adiponektyny 276 G/T i 11 377 C/G oraz rezystyny 420 C/G) w przypadku analizy pacjentek z nadwagą i otyłością z zespołem metabolicznym i pacjentek zdrowych z prawidłowym BMI. Uzyskane wyniki wskazują, że opisane powyżej polimorfizmy potencjalnie mogą wpływać na występowanie predyspozycji genetycznych w kierunku wystąpienia otyłości i zespołu metabolicznego. Nie stanowią one jednak czynnika różnicującego otyłość skojarzoną z zespołem metabolicznym od otyłości bez zespołu metabolicznego.
PODSUMOWANIE UZYSKANYCH WYNIKÓW
Celem przeprowadzonych badań była kompleksowa analiza zmian wydzielania adipokin i peptydów biorących udział w kontroli łaknienia u kobiet z różnym stopniem zaawansowania nadwagi i otyłości skojarzonej z zespołem metabolicznym w stosunku do analogicznych grup kobiet z BMI ≥ 25 kg/m2 bez towarzyszącego zespołu metabolicznego. Wszystkie wymienione poprzednio grupy kobiet z BMI ≥ 25 kg/m2 były także porównane do grupy kontrolnej młodych, szczupłych kobiet. Przedmiotem badań była również analiza częstości występowania polimorfizmów genów adiponektyny i rezystyny – dwóch adipokin wywierających przeciwstawne działania metaboliczne w rozwoju insulinoooporności. Oceniano także obecność ewentualnych asocjacji badanych polimorfizmów genów adiponektyny i rezystyny z występowaniem zespołu metabolicznego.
Na podstawie wykonanych badań podjęto próbę wyodrębnienia wskaźników peptydowych (wśród adipokin i peptydów biorących udział w kontroli łaknienia) oraz genetycznych (polimorfizmy genów adiponektyny i rezystyny) różnicujących osoby otyłe metabolicznie zdrowe (bez obecności zespołu metabolicznego) od osób otyłych ze współistniejącym zespołem metabolicznym.
Wyodrębnione peptydy i/lub czynniki genetyczne mogłyby być pomocne w wybraniu pacjentów wymagających szczególnie intensywnego nadzoru diagnostycznego i terapeutycznego ze względu na znacząco większe ryzyko wystąpienia powikłań sercowo-naczyniowych.
Spośród wszystkich badanych kobiet z nadwagą i otyłością szczególnie istotna była grupa kobiet z otyłością olbrzymią (BMI ≥ 40 kg/m 2), w której zespół metaboliczny pomimo skrajnie zaawansowanej otyłości występował jedynie w 73% przypadków.
Wykazanie w grupie kobiet z otyłością olbrzymią skojarzoną z zespołem metabolicznym różnic w profilu wydzielanych adipokin i zmian w częstości występowania badanych polimorfizmów genów adiponektyny i rezystyny w porównaniu do kobiet z BMI ≥ 40 kg/m 2 bez zespołu metabolicznego mogłoby w przyszłości skutkować wykorzystaniem badań stężeń adipokin oraz badań genetycznych w celu kwalifikowania chorych do kompleksowego leczenia otyłości, w tym zastosowania metod chirurgii bariatrycznej.
Należy podkreślić fakt, że nieprawidłowości biochemiczne lub zaburzenia peptydowe u osób otyłych mogą wyprzedzać wystąpienie klinicznych objawów takich chorób jak cukrzyca typu 2 i szeroko rozumiane choroby układu sercowo-naczyniowego.
Zakwalifikowane do badań kobiety podzielono na następujące grupy: I grupa – grupa kontrolna: kobiety z BMI <25 kg/m2; II grupa – kobiety z otyłością olbrzymią z BMI ≥ 40 kg/m2 (z obecnością i bez zespołu metabolicznego); III grupa – kobiety z nadwagą i otyłością z BMI 25-39,9 kg/m2 bez zespołu metabolicznego; IV grupa – kobiety z nadwagą i otyłością z BMI 25-39,9 kg/m2 z obecnością zespołu metabolicznego. U wszystkich badanych kobiet wykonano badania antropometryczne (BMI, BIA, WHR, obwód talii i bioder), pomiar ciśnienia tętniczego krwi, badania biochemiczne (lipidogram, stężenie glukozy i insuliny na czczo) oraz wyliczono wskaźnik HOMA-IR. Zespół metaboliczny rozpoznano na podstawie kryteriów IDF 2005.
Wśród badanych kobiet analizowano zmiany stężeń w surowicy następujących adipokin i peptydów: adiponektyny całkowitej i wysokocząsteczkowej, rezystyny, aktywnej greliny (ghrelin active), wisfatyny, leptyny, rozpuszczalnego receptora leptynowego oraz wyliczano indeks wolnej leptyny -FLI (free leptin index).
W badaniach genetycznych oznaczono 4 polimorfizmy genu adiponektyny i 3 polimorfizmy genu rezystyny. Analizę częstości występowania poszczególnych genotypów jak również ilorazu szans dla modelu dominującego przeprowadzono w stosunku do grupy kontrolnej kobiet z prawidłową masą ciała. Oceniano iloraz szans pomiędzy grupami pacjentek z nadwagą i otyłością z zespołem metabolicznym i bez zespołu metabolicznego. W celu zbadania ewentualnych asocjacji badanych polimorfizmów z otyłością i zespołem metabolicznym wykonano analizę regresji logistycznej
Analizowano także korelacje pomiędzy stężeniami adipokin w surowicy a wartościami wskaźników gospodarki lipidowej, węglowodanowej i wskaźnikami uzyskanymi w ocenie klinicznej pacjentek.
Wszystkie analizy statystyczne przeprowadzono zarówno w odniesieniu do ujednoliconej grupy kobiet z BMI> 25 kg/m2, jak i po rozdzieleniu badanych w na podgrupy (poszczególne analizowane grupy nadwagi i otyłości)
W wyniku przeprowadzonych badań stwierdzono:
1. Kobiety z otyłością olbrzymią miały najbardziej nasilone zmiany w profilu wydzielanych adipokin jak również wykazywały odmienne wartości korelacji pomiędzy badanymi adipokinami a wskaźnikami gospodarki lipidowej w porównaniu do kobiet z nadwagą i otyłością z BMI 25-39,9 kg/m2.
2. U kobiet z otyłością olbrzymią zanotowano najwyższe wartości leptyny, wisfatyny i rezystyny wśród analizowanych grup kobiet z nadwagą i otyłością oraz w stosunku do grupy kontrolnej.
3. Kobiety z otyłością olbrzymią miały najniższe wartości adiponektyny całkowitej i wysokocząsteczkowej oraz aktywnej greliny w surowicy wśród analizowanych grup kobiet z nadwagą i otyłością jak również w stosunku do grupy kontrolnej.
4. U pacjentek z otyłością olbrzymią wykazano odmienne niż w innych badanych grupach z nadwagą i otyłością korelacje pomiędzy leptyną a wskaźnikami gospodarki lipidowej (dodatnia korelacja leptyny z HDL-cholesterolem i ujemna z triglicerydami).
5. U kobiet z otyłością olbrzymią zanotowano też najsilniej wyrażony korzystny wpłwy wisfatyny na metabolizm cholesterolu (dodatnia korelacja wisfatyny z HDL-cholestreolem i ujemna z triglicerydami).
6. Po podzieleniu pacjentek z otyłością olbrzymią na grupę z obecnym zespołem metabolicznym i bez zespołu metabolicznego (metabolicznie zdrowych) stwierdzono znamiennie statystycznie wyższe wartości adiponektyny całkowitej i wisfatyny w grupie kobiet metabolicznie zdrowych. W grupie tej zanotowano także wyższe wartości HMW adiponektyny oraz niższe rezystyny w stosunku do kobiet z BMI ≥ 40 kg/m2 z towarzyszącym zespołem metabolicznym.
7. Porównanie grupy kobiet z BMI 25-39,9 kg/m2 z zespołem metabolicznym w stosunku do kobiet z BMI 25-39,9 kg/m2 bez zespołu metabolicznego wykazało znamiennie niższe wartości adiponektyny całkowitej i indeksu wolnej leptyny w grupie kobiet z BMI 25-39,9 kg/m2 skojarzonym z zespołem metabolicznym.
8. Stężenia adiponektyny całkowitej i HMW adiponektyny u kobiet z otyłością olbrzymią lub otyłością z BMI 25-39,9 kg/m2 bez towarzyszącego zespołu metabolicznego (metabolicznie zdrowych) były niższe niż u kobiet szczupłych z grupy kontrolnej, jednakże różnice te nie były istotne statystycznie.
9. Wykazano obecność istotnych korelacji pomiędzy stężeniami badanych adipokin a wartościami parametrów antropometrycznych i wskaźnikami metabolicznymi oraz insulinooporności we wszystkich badanych grupach kobiet z nadwagą i otyłością.
10. Obecność zespołu metabolicznego zarówno w przypadku otyłości z BMI 25-39,9 kg/m2 jak i otyłości olbrzymiej wiązała się z wyższymi wartościami ciśnienia tętniczego i gorszymi wskaźnikami gospodarki węglowodanowej.
11. Kobiety z nadwagą i otyłością w tym otyłością olbrzymią, którym towarzyszył zespół metaboliczny miały najwyższe wartości wskaźnika HOMA-IR oraz najsilniej wyrażone zaburzenia gospodarki lipidowej.
12. Przeprowadzone analizy wyników badań genetycznych pomiędzy pacjentkami z nadwagą i otyłością z lub bez zespołu metabolicznego nie wykazały istnienia istotnych statystycznie różnic pomiedzy powyższymi grupami.
13. W przypadku polimorfizmu 276 G/T genu adiponektyny stwierdzono, że obecność allelu T w genotypie pacjentek z otyłością olbrzymią lub nadwagą i otyłością (BMI 25-39,9 kg/m2) skojarzoną z zespołem metabolicznym może wiązać się z 2,5-krotnie mniejszym ryzykiem wystapienia otyłości olbrzymiej lub nadwagi i otyłości skojarzonej z zespołem metabolicznym w porównaniu do młodych szczupłych kobiet.
14. W zakresie polimorfizmu rezystyny 420 C/G obecność allelu G u pacjentek z nadwagą i otyłością z zespołem metabolicznym w stosunku do grupy szczupłych kobiet z prawidłowym BMI wiąże się z ponad 2 krotnym wzrostem ryzyka wystąpienia zespołu metabolicznego.
15. Wykazano możliwość asocjacji polimorfizmów adiponektyny 276 G/T, 11 377 C/G i rezystyny 420 C/G z zespołem metabolicznym analizując grupy pacjentek z nadwagą i otyłością z zespołem metabolicznym w stosunku do kobiet szczupłych.
Przedstawione wyniki badań kobiet z nadwagą i otyłością miały charakter kompleksowy ponieważ dokonano oceny wydzielania licznych adipokin i peptydów związanych z kontrolą łaknienia w odniesieniu do parametrów antropometrycznych i wyników badań biochemicznych. Badano także potencjalne asocjacje polimorfizmów genów adiponektyny i rezystyny w odniesieniu do otyłości i zespołu metabolicznego.
W wyniku przeprowadzonych badań stwierdzono znaczne różnice w profilu wydzielanych adipokin pomiędzy pacjentkami z nadwagą i otyłością skojarzoną z zespołem metabolicznym w odniesieniu do pacjentek z BMI ≥ 25 kg/m 2 bez zespołu metabolicznego (metabolicznie zdrowych). Wyodrębniono także adipokiny, które mogą być potencjalnym markerem zaburzeń metabolicznych w otyłości, co może mieć w przyszłości zastosowanie kliniczne.
WNIOSKI
1. Kobiety z otyłością bez zespołu metabolicznego (tzw. metabolicznie zdrowe) wykazywały istotne różnice w wydzielaniu adipokin w porównaniu do kobiet z otyłością skojarzoną z zespołem metabolicznym.
2. Pacjentki z otyłością olbrzymią skojarzoną z zespołem metabolicznym wykazywały najbardziej nasilone zaburzenia w wydzielaniu badanych adipokin.
3. Spośród badanych adipokin adiponektyna i jej frakcja wysokocząsteczkowa (HMW) wydają się być najlepszymi markerami zaburzeń metabolicznych w otyłości. Wyższe stężenia adiponektyny i jej frakcji HMW mogą być czynnikami zapobiegającymi wystąpieniu zespołu metabolicznego.
4. Współistnienie zespołu metabolicznego i towarzysząca mu insulinooporność wydaje się mieć większy wpływ na wydzielanie adipokin niż stopień klinicznego zaawansowania otyłości mierzony przy pomocy BMI oraz BIA.
5. Analizowane polimorfizmy genów adiponektyny i rezystyny nie wykazały asocjacji z nadwagą i otyłością skojarzoną z zespołem metabolicznym w odniesieniu do nadwagi i otyłości bez zespołu metabolicznego.
6. Badania peptydowe u osób z otyłością olbrzymią mogą być pomocne w podejmowaniu decyzji dotyczących kompleksowego leczenia tych osób a w szczególności rozważenia operacji bariatrycznej.
SPIS TREŚCI
Ocena zależności wydzielania adipokin i insulinooporności oraz asocjacji wybranych polimorfizmów genów adiponektyny i rezystyny w otyłości 912
WSTĘP 912
Otyłość – definicja i dane epidemiologiczne 912
Zespół metaboliczny 912
Tkanka tłuszczowa – narząd wydzielania wewnętrznego 913
Rola neuropeptydów wydzielanych w ośrodkowym układzie nerwowym i obwodowo w kontroli łaknienia 916
Zaburzenia hormonalne w otyłości 918
Czynniki genetyczne otyłości 919
UZASADNIENIE CELOWOŚCI PODJĘCIA BADAŃ 921
CEL PRACY 922
ZADANIA BADAWCZE 923
MATERIAŁ I METODY 924
ANALIZA STATYSTYCZNA 925
WYNIKI 926
DYSKUSJA I OMÓWIENIE UZYSKANYCH WYNIKÓW 948
Grupa kobiet z BMI ≥ 25 kg/m2 (otyłość olbrzymia, nadwaga i otyłość z lub bez zespołu metabolicznego – analiza łączna wszystkich grup) 948
Otyłość olbrzymia (BMI ≥ 40kg/m2) 952
Wyniki badań uzyskane u pacjentek z BMI ≥ 40 kg/m2 po wyodrębnieniu podgrup pacjentek z towarzyszącym zespołem metabolicznym i bez zespołu metabolicznego 955
Nadwaga i otyłość (BMI 25-39,9 kg/m2) z towarzyszącym zespołem metabolicznym 956
Nadwaga i otyłość (BMI 25-39,9 kg/m2) bez towarzyszącego zespołu metabolicznego 957
Porównanie wyników uzyskanych w grupach kobiet z BMI 25-39,9 z towarzyszącym zespołem metabolicznym i bez zespołu metabolicznego 957
Dyskusja wyników badań genetycznych 958
podsumowanie uzyskanych wyników 961
Wnioski 963
streszczenie 964
summary 970
piśmiennictwo 975
Podziękowanie
Praca została wykonana w Zakładzie Neuroendokrynologii Klinicznej CMKP w Warszawie oraz w Pracowni Biologii Molekularnej Kliniki Gastroenterologii i Hepatologii CMKP w Warszawie
Autor składa podziękowania za ogromną pomoc i życzliwość podczas realizacji kolejnych etapów badań:
? Wszystkim pracownikom Zakładu Neuroendokrynologii Klinicznej CMKP w Warszawie
? Prof. dr hab. Jerzemu Ostrowskiemu – Kierownikowi Pracowni Biologii Molekularnej Kliniki Gastroenterologii i Hepatologii CMKP oraz wszystkim pracownikom Pracowni Biologii Molekularnej
? Dyrekcji Centrum Medycznego Kształcenia Podyplomowego za stworzenie warunków do realizacji badań, których wyniki prezentowane są w tej pracy
Wojciech Bik
Piśmiennictwo
1. Definicja, epidemiologia i koszty otyłości. [W:] Tatoń J, Czech A, Bernas M: Otyłość. Zespół metaboliczny. Wydawnictwo Lekarskie PZWL, wyd. 1, 2007; s. 15-68.
2. Caro JF: Definitions and classification of obesity. Endotex.com. Chapter 2, 2002.
3. Must A, McKeown NM: The disease burden associated with overweight and obesity. Endotex.com. Chapter 2, 2002.
4. Ford ES, Mokdad AH: Epidemiology of obesity in the Western Hemisphere. J Clin Endocrinol Metab 2008; 93: S1-S8.
5. Misra A, Khurana L: Obesity and metabolic syndrome in developing countries J Clin Endocrinol Metab 2008; 93: S9-S30.
6. Bessesen DH: Update on obesity. J Clin Endocrinol Metab 2008; 93: 2027-2034.
7. Obuchowicz A: Epidemiologia nadwagi i otyłości – narastającego problemu zdrowotnego dzieci i młodzieży. Endokryn Otyłość i Zab Przem Materii 2005; 1: 9-12.
8. Wang Y, Monteiro C, Popkin BM: Trends of obesity and underweight in older children and adolescents in the United States, Brazil, China, and Russia. Am J Clin Nutr 2002; 75: 971-7.
9. Oblacińska A, Wrocławska M, Woynarowska B: Częstość występowania nadwagi i otyłości w populacji w wieku szkolnym w Polsce oraz opieka zdrowotna nad uczniami z tym zaburzeniem. Ped Pol 1997; 72: 241-45.
10. Smorczewska-Czupryńska M, Ustymowicz-Farbiszewska J, Karczewski J: Ocena występowania nadwagi i otyłości u dzieci szkół podstawowych Białegostoku i okolic. Przegl Ped 2000; 30: 303-6.
11. Lebiedowicz K, Staśkiewicz G, Torres K: Występowanie otyłości u 15-letnich dzieci w Lublinie w porównaniu z innymi krajami i czynnikami. Nowiny Lek 2001; 70: 55-60.
12. Milewicz A: Fenotypy otyłości a skład masy ciała i profil metaboliczny. Endokryn Otyłość i Zab Przem Materii 2005; 1: 15-19.
13. Must A, Jacques PF, Dallal GE et al.: Long-term morbidity and mortality of overweight adolescens a follow up of the Harvard Growth Study of 1922 to 1935. N Engl J Med 1992; 327: 1350-1355.
14. Cornier MA, Dabelea D, Hernandez TL et al.: The metabolic syndrome. Endocr Rev 2008; 29: 777-822.
15. Oda E: The metabolic syndrome as a concept of adipose tissue disease. Hypertens Res 2008; 31: 1283-91.
16. Kinalska I, Popławska-Kita A, Telejko B et al.: Otyłość a zaburzenia przemiany węglowodanowej. Endokryn Otyłość i Zab Przem Materii 2006; 2: 94-101.
17. Lakka HM, Lakka TA, Tuomilehto J et al.: Abdominal obesity is associated with increased risk of acute coronary events in men. Eur Heart J 2002; 23: 705-13.
18. Prineas RJ, Folsom AR, Kaye SA: Central adiposity and increased risk of coronary artery disease mortality in older women. Ann Epidemiol 1993; 3: 35-41.
19. Zhang Y, Proenca R, Maffei M et al.: Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372(6505): 425-32. Erratum in: Nature 1995; 374 (6521): 479.
20. Tartaglia LA, Dembski M, Weng X et al.: Identification and expression cloning of a leptin receptor, OB-R. Cell 1995; 83(7): 1263-71.
21. Siemińska L: Adipose tissue. Pathophysiology, distribution, sex differences and the role in inflammation and cancerogenesis Endokrynol Pol 2007; 58: 330-42.
22. Kershaw EE, Flier JS: Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 2004; 89: 2548-56.
23. Skowrońska B, Fichna M, Fichna P: Rola tkanki tłuszczowej w układzie dokrewnym. Endokryn Otyłość i Zab Przem Materii 2005; 1: 21-29.
24. Vázquez-Vela ME, Torres N, Tovar AR: White adipose tissue as endocrine organ and its role in obesity. Arch Med Res 2008; 39: 715-28.
25. Cannon B, Nedergaard J: Brown adipose tissue: function and physiological significance. Physiol Rev 2004; 84: 277-359.
26. Rabe K, Lehrke M, Parhofer KG et al.: Adipokines and insulin resistance. Mol Med 2008; 14: 745-51.
27. Unger RH: Minireview: weapons of lean body mass destruction: the role of ectopic lipids in metabolic syndrome. Endocrinology 2003; 144: 5159-5165.
28. Endokrynologia otyłości: wpływ systemowych regulacji endokrynnych na homeostazę energii. [W:] Tatoń J, Czech A, Bernas M: Otyłość. Zespół metaboliczny. Wydawnictwo Lekarskie PZWL, wyd. 1, 2007; s.148-167.
29. Otyłość brzuszna i zespół metaboliczny. [W:] Tatoń J, Czech A, Bernas M: Otyłość. Zespół metaboliczny. Wydawnictwo Lekarskie PZWL, wyd. 1, 2007; s. 191-224.
30. Patogeneza i diagnostyka insulinooporności. [W:] Tatoń J, Czech A, Bernas M: Diabetologia kliniczna. Wydawnictwo Lekarskie PZWL, wyd. 1, 2008; s. 162-172.
31. Karbowska J, Warczak E, Kochan Z: Polimorfizm genu i zaburzenia funkcjonalne adiponektyny jako jedna z przyczyn rozwoju oporności na insulinę. Postępy Hig Med Dośw 2004; 58: 449-57.
32. Meier U, Gressner AM: Endocrine regulation of energy metabolism: review of pathobiochemical and clinical chemical aspects of leptin, ghrelin, adiponectin and resitin. Clin Chem 2004; 50: 1511-25.
33. Karbowska J, Kochan Z: Role of adiponectin in the regulation of carbohydrate and lipid metabolism. J Phys Pharm 2006; 57, supp 6: 103-113.
34. Tilg H, Moschen AR: Role of adiponectin and PBEF/visfatin as regulators of inflammation: involvement in obesity-associated diseases. Clin Sci (Lond.) 2008; 114: 275-88.
35. Sattar N, Nelson SM: Adiponectin, diabetes and coronary heart disease in older person: unraveling the paradox. J Clin Endocr Metab 2008; 93: 3299-3301.
36. Dekker JM, Funahashi T, Nijpels G et al.: Prognostic value of adiponectin for cardiovascular disease and mortality. J Clin Endocrinol Metab 2008; 9: 1489-96.
37. Laoutaris ID, Vasiliadis IK, Dritsas A et al.: High plasma adiponectin is related to low functional capacity in patients with chronic heart failure. Int J Cardiol 2009 Jan 26. [Epub ahead of print].
38. Aguilar-Salinas CA, García EG, Robles L et al.: High adiponectin concentrations are associated with the metabolically healthy obese phenotype. J Clin Endocrinol Metab 2008; 93: 4075-9.
39. Arai Y, Nakazawa S, Kojima T et al.: High adiponectin concentration and its role for longevity in female centenarians. Geriatr Gerontol Int 2006; 6: 32-9.
40. Bik W, Baranowska-Bik A, Wolińska-Witort E et al.: The relationship between adiponectin levels and metabolic status in centenarian, early elderly, young and obese women. Neuro Endocrinol Lett 2006; 27: 493-500.
41. Atzmon G, Pollin TI, Crandall J et al.: Adiponectin levels and genotype: a potential regulator of life span in humans. J Gerontol A Biol Sci Med Sci 2008; 63: 447-53.
42. Tang NP, Wang LS, Yang L et al.: A polymorphism in the resistin gene promoter and the risk of coronary artery disease in a Chinese population. Clin Endocrinol (Oxf.) 2008; 68: 82-7.
43. Xu JY, Sham PC, Xu A et al.: Resistin gene polymorphisms and progression of glycaemia in southern Chinese: a 5-year prospective study. Clin Endocrinol (Oxf.) 2007; 66: 211-7.
44. Banerjee RR, Lazar MA: Resistin: molecular history and prognosis. J Mol Med 2003; 81: 218-26.
45. Steppan CM, Bailey ST, Bhat S et al.: The hormone resistin links obesity to diabetes. Nature 2001; 409: 307-12.
46. Hivert MF, Sullivan LM, Fox CS et al.: Associations of adiponectin, resistin, and tumor necrosis factor-alpha with insulin resistance. J Clin Endocrinol Metab 2008; 93: 3165-72.
47. Lehrke M, Reilly MP, Millington SC et al.: An inflammatory cascade leading to hyperresistinemia in humans. PLoS Med 2004; 1(2): e45; 161-168.
48. Pang SS, Le YY: Role of resistin in inflammation and inflammation-related diseases. Cell Mol Immunol 2006; 3(1): 29-34.
49. Adeghate E: Visfatin: structure, function and relation to diabetes mellitus and other dysfunctions. Curr Med Chem 2008; 15: 1851-62.
50. Körner A, Garten A, Blüher M et al.: Molecular characteristics of serum visfatin and differential detection by immunoassays. J Clin Endocrinol Metab 2007; 92(12): 4783-91.
51. Bełtowski J: Apelin and visfatin: unique „beneficial” adipokines upregulated in obesity? Med Sci Monit 2006; 12(6): RA112-9.
52. Tokunaga A, Miura A, Okauchi Y et al.: The-1535 promoter variant of the visfatin gene is associated with serum triglyceride and HDL-cholesterol levels in Japanese subjects. Endocr J 2008; 55(1): 205-12.
53. Johansson LM, Johansson LE, Ridderstrale M: The visfatin (PBEF1) G-948T gene polymorphism is associated with increased high-density lipoprotein cholesterol in obese subjects. Metabolism 2008; 57: 1558-62.
54. Malyszko J, Malyszko JS, Pawlak K et al.: Visfatin and apelin, new adipocytokines, and their relation to endothelial function in patients with chronic renal failure. Adv Med Sci 2008; 53: 32-6.
55. Dahmen N, Manderscheid N, Helfrich J et al.: Elevated peripheral visfatin levels in narcoleptic patients. PLoS ONE 2008; 3: e2980.
56. Walczewska A: Leptyna nowy hormone. Pol J Endocrinol 2000; 51: 125-148.
57. Rasouli N, Kern PA: Adipokines and metabolic complications of obesity. J Clin Endocrinol Metab 2008; 93: S64-S73.
58. Baranowska B, Wolinska-Witort E, Wasilewska-Dziubinska E et al.: Plasma leptin, neuropeptide Y (NPY) and galanin concentrations in bulimia nervosa and in anorexia nervosa. Neuro Endocrinol Lett 2001; 22: 356-8.
59. Kaye W: Neurobiology of anorexia and bulimia nervosa. Physiol Behav 2008; 94: 121-135.
60. Södersten P, Nergardh R, Bergh C et al.: Behavioral neuroendocrinology and treatment of anorexia nervosa. Front Neuroendocrinol 2008; 29: 445-62.
61. Zhang Y, Scarpace PJ: The role of leptin in leptin resistance and obesity. Physiol Behav 2006; 88: 249-56.
62. Banks WA, Farr SA, Morley JE: The effects of high fat diets on the blood-brain barrier transport of leptin: failure or adaptation. Physiol Behav 2006; 88: 244-48.
63. Faggioni R, Fantuzzi G, Gabay C et al.: Leptin deficiency enhances sensitivity to endotoxin-induced lethality. Am J Physiol 1999; 276: R136-42.
64. Sarraf P, Frederich RC, Turner EM et al.: Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. J Exp Med 1997; 185: 171-5.
65. Lord GM: Leptin as a proinflammatory cytokine. Contrib Nephrol 2006; 151: 151-64.
66. Lord GM, Matarese G, Howard JK et al.: Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 1998; 394: 897-901.
67. Beltowski J: Leptin and atherosclerosis. Atherosclerosis. 2006; 189: 47-60.
68. Hajer GR, van Haeften TW, Visseren FL: Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur Heart J 2008; 29: 2959-71.
69. Baranowska B: Neuroendokrynna kontrola głodu i sytości. [W:] Choroby wewnętrzne. Pod red.: A. Szczeklika, t. I, wyd. I, MP, s.1217-1218.
70. Stawiarska-Pięta B, Paszczela A, Szafarska-Stojko E: Patofizjologia otyłości – zaburzenia mechanizmów regulacji łaknienia w aspekcie otyłości. Ann Acad Med Siles 2007; 61: 77-88.
71. Arora S, Anubhuti: Role of neuropeptides in apeptite regulation and obesity – A reveiw Neuropeptides 2006; 40: 371-401.
72. Crowell MD, Decker GA, Levy R et al.: Gut-brain neuropeptides in the regulation of ingestive behaviors and obesity. Am J Gastroenterol 2006; 101: 2848-56.
73. Kalra SP, Kalra PS: NPY and cohorts in regulating appetite, obesity and metabolic syndrome: beneficial effects of gene therapy. Neuropeptides 2004; 38: 201-11.
74. Inui A: Transgenic approach to the study of body weight regulation. Pharmacol Rev 2000; 52: 35-62.
75. Yang YK, Thompson DA, Dickinson CJ et al.: Characterization of Agouti-related protein binding to melanocortin receptors. Mol Endocrinol 1999; 13: 148-55.
76. Yang YK, Dickinson CJ, Zeng Q et al.: Contribution of melanocortin receptor exoloops to Agouti-related protein binding. J Biol Chem 1999; 274: 14100-6.
77. Elias CF, Lee CE, Kelly JF et al.: Characterization of CART neurons in the rat and human hypothalamus. J Comp Neurol 2001; 432: 1-19.
78. Li HY, Hwang HW, Hu YH: Functional characterizations of cocaine- and amphetamine-regulated transcript mRNA expression in rat hypothalamus. Neurosci Lett 2002; 323: 203-6.
79. Baranowska B, Wolińska-Witort E, Martyńska L et al.: Effects of cocaine-amphetamine regulated transcript (CART) on hormone release. Regul Pept 2004; 122: 55-9.
80. Baranowska B, Wolinska-Witort E, Bik W et al.: The effect of cocaine-amphetamine regulated transcript (CART) on the activation of the pituitary-adrenal axis. Neuro Endocrinol Lett 2006; 27: 60-2.
81. Larsen PJ, Vrang N, Petersen PC et al.: Chronic intracerebroventricular administration of recombinant CART(42-89) peptide inhibits and causes weight loss in lean and obese Zucker (fa/fa) rats. Obes Res 2000; 8: 590-6.
82. Bik W, Skwarlo-Sonta K, Szelagiewicz J et al.: Involvement of the cocaine-amphetamine regulated transcript peptide (CART 55-102) in the modulation of rat immune cell activity. Neuro Endocrinol Lett 2008; 29: 359-65.
83. Pritchard LE, Turnbull AV, White A: Pro-opiomelanocortin processing in the hypothalamus: impact on melanocortin signalling and obesity. J Endocrinol 2002; 172: 411-21.
84. Pritchard LE, Oliver RL, McLoughlin JD et al.: Proopiomelanocortin-derived peptides in rat cerebrospinal fluid and hypothalamic extracts: evidence that secretion is regulated with respect to energy balance. Endocrinology 2003; 144: 760-6.
85. Neary NM, Goldstone AP, Bloom SR: Appetite regulation: from the gut to the hypothalamus.Clin Endocrinol (Oxf.) 2004; 60: 153-60.
86. Hagan MM, Rushing PA, Pritchard LM et al.: Long-term orexigenic effects of AgRP-(83-132) involve mechanisms other than melanocortin receptor blockade. Am J Physiol Regul Integr Comp Physiol 2000; 279: R47-52.
87. Rossi M, Kim MS, Morgan DG et al.: A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology 1998; 139: 4428-31.
88. Fu LY, van den Pol AN: Agouti-related peptide and MC3/4 receptor agonists both inhibit excitatory hypothalamic ventromedial nucleus neurons. J Neurosci 2008; 28: 5433-49.
89. Hoch M, Hirzel E, Lindinger P et al.: Weak functional coupling of the melanocortin-1 receptor expressed in human adipocytes. J Recept Signal Transduct Res 2008; 28: 485-504.
90. Zobel DP, Andreasen CH, Grarup N et al.: Variants near MC4R associate with obesity and influence obesity-related quantitative traits in a population of middle-aged people: studies of 14,940 Danes. Diabetes 2009; 58: 757-64.
91. Yurtcu E, Yilmaz A, Ozkurt Z et al.: Melanocortin-4 Receptor Gene Polymorphisms in Obese Patients. Biochem Genet 2009; 47: 295-300.
92. Jesudason D, Wittert G: Endocannabinoid system in food intake and metabolic regulation. Curr Opin Lipidol 2008; 19: 344-8.
93. Kozakowski J, Zgliczyński W: Rola układu endokannabinoidowego w patogenezie otyłości: Post Nauk Med 2008; 21: 198-202.
94. Cota D, Marsicano G, Tschöp M et al.: The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest 2003; 112: 423-31.
95. Di Marzo V, Capasso R, Matias I et al.: The role of endocannabinoids in the regulation of gastric emptying: alterations in mice fed a high-fat diet. Br J Pharmacol 2008; 153: 1272-80.
96. Di Marzo V, Goparaju SK, Wang L et al.: Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001; 410(6830): 822-5.
97. Katergari SA, Milousis A, Pagonopoulou O et al.: Ghrelin in pathological conditions. Endocr J 2008; 55: 439-53.
98. Kojima M, Hosoda H, Date Y et al.: Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999; 402: 656-60.
99. Arvat E, Maccario M, Di Vito L et al.: Endocrine activities of ghrelin, a natural growth hormone secretagogue (GHS), in humans: comparison and interactions with hexarelin, a nonnatural peptidyl GHS, and GH-releasing hormone. J Clin Endocrinol Metab 2001; 86: 1169-74.
100. Nakai Y, Hosoda H, Nin K et al.: Plasma levels of active form of ghrelin during oral glucose tolerance test in patients with anorexia nervosa.. Eur J Endocrinol 2003; 149: R1-3.
101. Hellerstein MK, Parks EJ: Otyłość i nadwaga. [W:] Endokrynologia ogólna i kliniczna. Red.: FS Greenspan, DG Gardner. Wyd. Czelej, Lublin 2004; 800-819.
102. Demissie M, Milewicz A: Zaburzenia hormonalne w otyłości. Diabetol Prakt 2003; 4: 207-209.
103. Baranowska B, Wolińska-Witort E, Martyńska L et al.: Sibutramine therapy in obese women-effects on plasma neuropeptide Y (NPY), insulin, leptin and beta-endorphin concentrations. Neuro Endocrinol Lett 2005; 26: 675-9.
104. García de la Torre N, Rubio MA, Bordiú E et al.: Effects of weight loss after bariatric surgery for morbid obesity on vascular endothelial growth factor-A, adipocytokines, and insulin. J Clin Endocrinol Metab 2008; 93: 4276-81.
105. Pasquali R, Vicennati V, Cacciami M et al.: The hypothalamic-pituitary-adrenal axis activity in obesity and metabolic syndrome. Ann NY Acad Sci 2006; 1083: 111-28.
106. Salehi M, Ferenczi A, Zumoff B: Obesity and cortisol status. Horm Metab Res 2005; 37: 193-7.
107. Wauters M, Considine RV, Gaal van LF: Human leptin: from adipocyte hormone to an endocrine mediator. Eur J Endocrinol 2000; 143: 293-311.
108. Widjaja A, Schurmeyer TH, von zur Muchlen A et al.: Determinants of serum leptin levels in Cushing´s syndrome. J Clin Endocrinol Metab 1998; 83: 600-3.
109. Franco C, Bengtsson BA, Johannsson G: The GH/IGF1 axis in obesity: physiological and pathological aspects. Metab Syndr Relat Disord 2006; 4: 51-6.
110. Johannsson G, Bengtsson BA: Growth hormone and metabolic syndrome. J Endocrinol Invest 1999; 22(5 Suppl): 41-6.
111. Johansen T, Malmlöf K: Treatment of obesity using GH. Metab Syndr Relat Disord 2006; 4: 59-69.
112. Portmann L, Giusti V: Obesity and hypothyroidism: myth or reality? Rev Med Suisse 2007; 3: 859-62.
113. Orban Z, Bornstein SR, Chrousos GP: The interaction between leptin and the hypothalamic-pituitary-thyroid axis. Horm Metab Res 1998; 30: 231-5.
114. Kotulska A, Kucharz EJ: Leptyna a hormony tarczycy. Przegl Lek 2002; 59: 1024-7.
115. Tatoń J, Czech A, Bernas M: Genetyczne podstawy etiologii otyłości. [W:] Otyłość. Zespół metaboliczny. Wydawnictwo Lekarskie PZWL, wyd. 1, 2007; 69-96.
116. Blakemore AIF, Froguel P: Is obesity our genetic legacy? J Clin Endocrinol Metab 2008; 93: S51-S56.
117. Walley AJ, Blakemore AIF, Froguel P: Genetics of obesity and the prediction of risk for health. Hum Mol Gen 2006; 15: R124-R130.
118. Barness LA, Opiz JM, Gilbert-Barness E: Obesity: Genetic, molecular and environmental aspects. Am J Med Genet Part A 2007; 143A: 3016-3034.
119. Prentice AM: Early influences on human energy regulation: thrifty genotypes and thrifty phenotypes. Physiol Behav 2005; 86: 640-5.
120. Lusis AJ, Attie AD, Reje K: Metabolic syndrome: from epidemiology to systemic biology. Nat Rev Genet 2008; 9: 819-30.
121. Krzyżanowska-Świniarska B, Koziołek M: Otyłość. Zespół metaboliczny. [W:] Endokrynologia w codziennej praktyce lekarskiej. Red.: A. Syrenicz. Wyd. Pomorskiej Akademii Medycznej w Szczecinie, s. 647-667.
122. Wardle J, Carnell S, Haworth CMA et al.: Obesity associated genetic variation in FTO is associated with diminished satiety. J Clin Endocrinol Metab 2008; 93: 3640-43.
123. Cecil JE, Tavendale R, Watt P et al.: An obesity-associated FTO gene variant and increased energy intake in children. N Engl J Med 2008; 359: 2603-4.
124. Müller TD, Hinney A, Scherag A et al.: Fat mass and obesity associated´ gene (FTO): no significant association of variant rs9939609 with weight loss in a lifestyle intervention and lipid metabolism markers in German obese children and adolescents. BMC Med Genet 2008; 9: 85.
125. Gerken T, Girard CA, Tung YC et al.: The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 2007; 318: 1469-72.
126. Haupt A, Thamer C, Staiger H et al.: Variation in the FTO Gene Influences Food Intake but not Energy Expenditure. Exp Clin Endocrinol Diabetes 2009; 117: 194-7.
127. Vidal-Puig A, Jimenez-Lińan M, Lowell BB et al.: Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J Clin Invest 1996; 97: 2553-61.
128. Cecil JE, Watt P, Palmer CN et al.: Energy balance and food intake. The role of PPARγ polymorphisms 2006; 88: 227-233.
129. Kadowaki T, Yamauchi T, Kubota N et al.: Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 2006; 116: 1784-92.
130. Yang WS, Chuang LM: Human genetics of adiponectin in the metabolic syndrome. J Mol Med 2006; 84: 112-21.
131. Joy T, Lahiry P, Pollex RL et al.: Genetics of metabolic syndrome.Curr Diab Rep 2008; 8: 141-8.
132. Szopa M, Malczewska-Malec M, Wilk B et al.: Variants of the adiponectin gene and type 2 diabetes in a Polish population. Acta Diabetol 2009, Jan 28. [Epub ahead of print].
133. Rasmussen-Torvik LJ, Pankow JS, Jacobs DR Jr et al.: The association of SNPs in ADIPOQ, ADIPOR1, and ADIPOR2 with insulin sensitivity in a cohort of adolescents and their parents. Hum Genet 2009; 125: 21-8.
134. Wang H, Chu WS, Hemphil CH et al.: Human resistin gene: molecular scanning and evaluation of association with insulin sensitivity and type 2 diabetes in Caucasians. J Clin Endocrinol Metab 2002; 87: 2520-2524.
135. Tan MS, Chang SY, Chang DM et al.: Association of resistin gene 3´-untranslated region +62G->A polymorphism with type 2 diabetes and hypertension in a Chinese population. J Clin Endocrinol Metab 2003; 88: 1258-63.
136. Gouni-Berthold I, Giannakidou E, Faust M et al.: Resistin gene 3´-untranslated region +62G->A polymorphism is associated with hypertension but not diabetes mellitus type 2 in a German population. J Intern Med 2005; 258: 518-26.
137. Dyslipidemia cukrzycowa: diagnostyka i leczenie. [W:] Tatoń J, Czech A, Bernas M: Diabetologia Kliniczna. wyd. 1, Wydawnictwo Lekarskie PZWL 2008; 580-595.
138. Aguilera CM, Gil-Campos M, Cańete R et al.: Alterations in plasma and tissue lipids associated with obesity and metabolic syndrome. Clin Sci (Lond.) 2008; 114: 183-93.
139. Brahami-Horn MC, Pouyssegur J: Oxygen a source of life and stress. FEBS Lett 2007; 294: E336-E344.
140. Lumenng CN, Bodzin JL, Saltiel AR: Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007; 117: 175-184.
141. Gordon S, Taylor PR: Monocyte and macrophage heterogeneity. Nat Rev Immunnol 2005; 5: 953-964.
142. Permana PA, Menge C, Reaven PD: Macrophage-secreted factors induce adipocyte inflammation and insulin resistance. Biochem Biophys Res Commun 2006; 341: 507-14.
143. Ruan H, Hacohen N, Golub TR et al.: Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: nuclear factor-kappaB activation by TNF-alpha is obligatory. Diabetes 2002; 51: 1319-36.
144. Tsigos C, Papanicolaou DA, Kyrou I et al.: Dose-dependent effects of recombinant human interleukin-6 on glucose regulation. J Clin Endocrinol Metab 1997; 82: 4167-70.
145. Rega G, Kaun C, Demyanets S et al.: Vascular endothelial growth factor is induced by the inflammatory cytokines interleukin-6 and oncostatin m in human adipose tissue in vitro and in murine adipose tissue in vivo. Arterioscler Thromb Vasc Biol 2007; 27: 1587-95.
146. Kadowaki T, Yamauchi T, Kubota N: The physiological and pathophysiological role of adiponectin and adiponectin receptors in the peripheral tissues and CNS. FEBS Lett 2008; 582: 74-80.
147. O´Rourke L, Yeaman SJ, Shepherd PR: Insulin and leptin acutely regulate cholesterol ester metabolism in macrophages by novel signaling pathways. Diabetes 2001; 50: 955-61.
148. Böger RH, Bode-Böger SM, Frölich JC: The L-arginine-nitric oxide pathway: role in atherosclerosis and therapeutic implications. Atherosclerosis 1996; 127: 1-11.
149. Mastronardi CA, Yu WH, McCann SM: Resting and circadian release of nitric oxide is controlled by leptin in male rats. Proc Natl Acad Sci USA 2002; 99: 5721-6.
150. Kimura K, Tsuda K, Baba A et al.: Involvement of nitric oxide in endothelium-dependent arterial relaxation by leptin. Biochem Biophys Res Commun 2000; 273: 745-9.
151. Vecchione C, Maffei A, Colella S et al.: Leptin effect on endothelial nitric oxide is mediated through Akt-endothelial nitric oxide synthase phosphorylation pathway. Diabetes 2002; 5: 168-73.
152. Söderberg S, Stegmayr B, Ahlbeck-Glader C et al.: High leptin levels are associated with stroke. Cerebrovasc Dis 2003; 15: 63-9.
153. Trujillo ME, Scherer PE: Adipose tissue-derived factors: impact on health and disease. Endocr Rev 2006; 27: 762-78.
154. Trayhurn P, Wood IS: Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr 2004; 92: 347-55.
155. Kadowaki T, Yamauchi T: Adiponectin and adiponectin receptors. Endocr Rev 2005; 26: 439-51.
156. Zhu W, Cheng KK, Vanhoutte PM et al.: Vascular effects of adiponectin: molecular mechanisms and potential therapeutic intervention. Clin Sci (Lond.) 2008; 114: 361-74.
157. Pischon T, Girman CJ, Hotamisligil GS et al.: Plasma adiponectin levels and risk of myocardial infarction in men. JAMA 2004; 291: 1730-7.
158. Schulze MB, Shai I, Rimm EB et al.: Adiponectin and future coronary heart disease events among men with type 2 diabetes. Diabetes 2005; 54: 534-9.
159. McEntegart MB, Awede B, Petrie MC et al.: Increase in serum adiponectin concentration in patients with heart failure and cachexia: relationship with leptin, other cytokines, and B-type natriuretic peptide. Eur Heart J 2007; 28: 829-35.
160. Kubota N, Yano W, Kubota T et al.: Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake.Cell Metab 2007; 6: 55-68.
161. Vasseur F, Helbecque N, Dina C et al.: Single-nucleotide polymorphism haplotypes in the both proximal promoter and exon 3 of the APM1 gene modulate adipocyte-secreted adiponectin hormone levels and contribute to the genetic risk for type 2 diabetes in French Caucasians. Hum Mol Genet 2002; 11: 2607-14.
162. Waki H, Yamauchi T, Kamon J et al.: Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J Biol Chem 2003; 278: 40352-63.
163. Haqq AM, Muehlbauer M, Svetkey LP et al.: Altered distribution of adiponectin isoforms in children with Prader-Willi syndrome (PWS): association with insulin sensitivity and circulating satiety peptide hormones. Clin Endocrinol (Oxf.) 2007; 67: 944-51.
164. Kaser S, Tatarczyk T, Stadlmayr A et al.: Effect of obesity on insulin sensitivity on adiponectin isoform distribution. Eur J Clin Invest 2008; 38: 827-34.
165. Polak J, Kovacova Z, Holst C et al.: Total adiponectin and adiponectin multimeric complexes in relation to weight loss-induced improvements in insulin sensitivity in obese women: the NUGENOB study. Eur J Endocrinol 2008; 158: 533-41.
166. Kovacova Z, Vitkova M, Kovacikova M et al.: Secretion of adiponectin multimeric complexes from adipose tissue explants is not modified by very low calorie diet. Eur J Endocrinol 2009; 160: 585-92.
167. O´Leary VB, Jorett AE, Marchetti CM et al.: Enhanced adiponectin multimer ratio and skeletal muscle adiponectin receptor expression following exercise training and diet in older insulin-resistant adults. Am J Physiol Endocrinol Metab 2007; 293: E421-7.
168. Lara-Castro C, Fu Y, Chung BH et al.: Adiponectin and the metabolic syndrome: mechanisms mediating risk for metabolic and cardiovascular disease. Curr Opin Lipidol 2007; 18: 263-70.
169. Lara-Castro C, Luo N, Wallace P et al.: Adiponectin multimeric complexes and the metabolic syndrome trait cluster. Diabetes 2006; 55: 249-59.
170. Sattar N, Watt P, Cherry L et al.: High molecular weight adiponectin is not associated with incident coronary heart disease in older women: a nested prospective case-control study. J Clin Endocrinol Metab 2008; 93: 1846-9.
171. Yano Y, Toshinai K, Inokuchi T et al.: Plasma des-acyl ghrelin, but not plasma HMW adiponectin, is a useful cardiometabolic marker for predicting atherosclerosis in elderly hypertensive patients. Atherosclerosis 2009; 204: 590-4.
172. Gualillo O, González-Juanatey JR, Lago F: The emerging role of adipokines as mediators of cardiovascular function: physiologic and clinical perspectives. Trends Cardiovasc Med 2007; 17: 275-83.
173. Rajala MW, Obici S, Scherer PE et al.: Adipose-derived resistin and gut-derived resistin-like molecule-beta selectively impair insulin action on glucose production. J Clin Invest 2003; 111: 225-30.
174. Curat CA, Wegner V, Sengenes C et al.: Macrophages in human visceral adipose tissue: increased accumulation in obesity and a source of resistin and visfatin. Diabetologia 2006; 49: 744-7.
175. Sundén-Cullberg J, Nyström T, Lee ML et al.: Pronounced elevation of resistin correlates with severity of disease in severe sepsis and septic shock. Crit Care Med 2007; 35: 1536-42.
176. Migita K, Maeda Y, Miyashita T et al.: The serum levels of resistin in rheumatoid arthritis patients. Clin Exp Rheumatol 2006; 24: 698-701.
177. Senolt L, Housa D, Vernerová Z et al.: Resistin in rheumatoid arthritis synovial tissue, synovial fluid and serum. Ann Rheum Dis 2007; 66: 458-63.
178. Barazzoni R, Zanetti M, Ferreira C et al.: Relationships between desacylated and acylated ghrelin and insulin sensitivity in the metabolic syndrome. J Clin Endocrinol Metab 2007; 92: 3935-40.
179. Siejka A, Ruxer J, Loba J: Ghrelin-role in energy homeostasis and glucose metabolism Endokrynol Diabetol Chor Przemiany Materii Wieku Rozw 2005; 11: 181-5.
180. Zwirska-Korczala K, Konturek SJ, Sodowski M et al.: Basal and postprandial plasma levels of PYY, ghrelin, cholecystokinin, gastrin and insulin in women with moderate and morbid obesity and metabolic syndrome. J Physiol Pharmacol 2007; 58, Suppl 1: 13-35.
181. Baranowska B, Bik W, Baranowska-Bik A et al.: Neuroendocrine control of metabolic homeostasis in Polish centenarians. J Physiol Pharmacol 2006; 57 Suppl 6: 55-61.
182. Patel JV, Cummings DE, Girod JP et al.: Role of metabolically active hormones in the insulin resistance associated with short-term glucocorticoid treatment. J Negat Results Biomed 2006; 5: 14.
183. Tanaka M, Nozaki M, Fukuhara A et al.: Biochem Visfatin is released from 3T3-L1 adipocytes via a non-classical pathway. Biophys Res Commun 2007; 359: 194-201.
184. Ognjanovic S, Bao S, Yamamoto SY et al.: Genomic organization of the gene coding for human pre-B-cell colony enhancing factor and expression in human fetal membranes. J Mol Endocrinol 2001; 26: 107-17.
185. Kralisch S, Klein J, Lossner U et al.: Interleukin-6 is a negative regulator of visfatin gene expression in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 2005; 289: E586-90.
186. Poulain-Godefroy O, Froguel P: Preadipocyte response and impairment of differentiation in an inflammatory environment. Biochem Biophys Res Commun 2007; 356: 662-7.
187. Haider DG, Schaller G, Kapiotis S et al.: The release of the adipocytokine visfatin is regulated by glucose and insulin. Diabetologia 2006; 49: 1909-14.
188. Sinha MK, Songer T, Xiao Q et al.: Analytical validation and biological evaluation of a high molecular-weight adiponectin ELISA. Clin Chem 2007; 53: 2144-51.
189. Leung KC, Xu A, Craig ME et al.: Adiponectin isoform distribution in women-relationship to femal sex, steroids and insulin sensitivity Metabolism 2009; 58: 239-45.
190. Salani B, Briatore L, Andraghetti G et al.: High-molecular weight adiponectin isoforms increase after biliopancreatic diversion in obese subjects. Obesity (Silver Spring) 2006; 14: 1511-4.
191. Swarbrick MM, Austrheim-Smith IT, Stanhope KL et al.: Circulating concentrations of high-molecular-weight adiponectin are increased following Roux-en-Y gastric bypass surgery. Diabetologia 2006; 49: 2552-8.
192. Swarbrick MM, Stanhope KL, Austrheim-Smith IT et al.: Longitudinal changes in pancreatic and adipocyte hormones following Roux-en-Y gastric bypass surgery. Diabetologia 2008; 51: 1901-11.
193. Engl J, Bobbert T, Ciardi C et al.: Effects of pronounced weight loss on adiponectin oligomer composition and metabolic parameters. (Silver Spring) 2007; 15: 1172-8.
194. Bełtowski J: Role of leptin in blood pressure regulation and arterial hypertension. J Hypertens 2006; 24: 789-801.
195. Ingelsson E, Larson MG, Yin X et al.: Circulating ghrelin, leptin, and soluble leptin receptor concentrations and cardiometabolic risk factors in a community-based sample. J Clin Endocrinol Metab 2008; 93: 3149-57.
196. Thomopoulos C, Papadopoulos DP, Papazachou O et al.: Free Leptin Is Associated With Masked Hypertension in Nonobese Subjects. A Cross-Sectional Study. Hypertension 2009; 53: 965-72.
197. Lembo G, Vecchione C, Fratta L et al.: Leptin induces direct vasodilation through distinct endothelial mechanisms. Diabetes 2000; 49: 293-7.
198. Romero-Corral A, Sierra-Johnson J, Lopez-Jimenez F et al.: Relationships between leptin and C-reactive protein with cardiovascular disease in the adult general population. Nat Clin Pract Cardiovasc Med 2008; 5: 418-25.
199. Sung SH, Chuang SY, Sheu WH et al.: Adiponectin, but not leptin or high-sensitivity C-reactive protein, is associated with blood pressure independently of general and abdominal adiposity. Hypertens Res 2008; 31: 633-40.
200. Adami GF, Civalleri D, Cella F et al.: Relationships of serum leptin to clinical and anthropometric findings in obese patients. Obes Surg 2002; 12: 623-7.
201. García-Lorda P, Bulló M, Vilŕ R et al.: Leptin concentrations do not correlate with fat mass nor with metabolic risk factors in morbidly obese females. Diabetes Nutr Metab 2001; 14: 329-36.
202. Lee Y, Wang MY, Kakuma T et al.: Liporegulation in diet-induced obesity. The antisteatotic role of hyperleptinemia. J Biol Chem 2001; 276: 5629-35.
203. Steinberg GR, Parolin ML, Heigenhauser GJ et al.: Leptin increases FA oxidation in lean but not obese human skeletal muscle: evidence of peripheral leptin resistance. Am J Physiol Endocrinol Metab 2002; 283: E187-92.
204. Unger RH: Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol Metab 2003; 14: 398-403.
205. Sinha MK, Opentanova I, Ohannesian JP et al.: Evidence of free and bound leptin in human circulation. Studies in lean and obese subjects and during short-term fasting. J Clin Invest 1996; 98: 1277-82.
206. Landt M: Leptin binding and binding capacity in serum. Clin Chem 2000; 46: 379-84.
207. Lammert A, Kiess W, Bottner A et al.: Soluble leptin receptor represents the main leptin binding activity in human blood. Biochem Biophys Res Commun 2001; 283: 982-8.
208. Haider DG, Schindler K, Schaller G et al.: Increased plasma visfatin concentrations in morbidly obese subjects are reduced after gastric banding. J Clin Endocrinol Metab 2006; 91: 1578-81.
209. Botella-Carretero JI, Luque-Ramírez M, Alvarez-Blasco F et al.: The increase in serum visfatin after bariatric surgery in morbidly obese women is modulated by weight loss, waist circumference, and presence or absence of diabetes before surgery. Obes Surg 2008; 18: 1000-6.
210. García-Fuentes E, García-Almeida JM, García-Arnés J et al.: Plasma visfatin concentrations in severely obese subjects are increased after intestinal bypass. Obesity (Silver Spring) 2007; 15: 2391-5.
211. Krzyżanowska K, Mittermayer F, Krugluger W et al.: Increase in visfatin after weight loss induced by gastroplastic surgery. Obesity (Silver Spring) 2006; 14: 1886-9.
212. Wang P, van Greevenbroek MM, Bouwman FG et al.: The circulating PBEF/NAMPT/visfatin level is associated with a beneficial blood lipid profile. Pflugers Arch 2007; 454: 971-6.
213. Smith J, Al-Amri M, Sniderman A et al.: Visfatin concentration in Asian Indians is correlated with high density lipoprotein cholesterol and apolipoprotein A1. Clin Endocrinol (Oxf.) 2006; 65: 667-72.
214. Jin H, Jiang B, Tang J et al.: Serum visfatin concentrations in obese adolescents and its correlation with age and high-density lipoprotein cholesterol. Diabetes Res Clin Pract 2008; 79: 412-8.
215. Dahl TB, Yndestad A, Skjelland M et al.: Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: possible role in inflammation and plaque destabilization. Circulation 2007; 115: 972-80.
216. Archer SL: Pre-B-cell colony-enhancing factor regulates vascular smooth muscle maturation through a NAD+-dependent mechanism: recognition of a new mechanism for cell diversity and redox regulation of vascular tone and remodeling. Circ Res 2005; 97: 4-7.
217. Takahashi Y, Tanaka A, Nakamura T et al.: Nicotinamide suppresses hyperphosphatemia in hemodialysis patients. Kidney Int 2004; 65: 1099-104.
218. Karelis AD: Metabolically healthy but obese individuals. Lancet 2008; 372(9646): 1281-3.
219. Marini MA, Succurro E, Frontoni S et al.: Metabolically healthy but obese women have an intermediate cardiovascular risk profile between healthy nonobese women and obese insulin-resistant women. Diabetes Care 2007; 30: 2145.
220. Francischetti EA, Celoria BM, Duarte SF et al.: Hypoadiponectinemia is associated with blood pressure increase in obese insulin-resistant individuals. Metabolism 2007; 56: 1464-9.
221. Kern PA, Ranganathan S, Li C et al.: Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab 2001; 280: E745-51.
222. Uysal KT, Wiesbrock SM, Hotamisligil GS: Functional analysis of tumor necrosis factor (TNFα) receptors in TNF-alpha-mediated insulin resistance in genetic obesity. Endocrinology 1998; 139: 4832-8.
223. Ouchi N, Kihara S, Arita Y et al.: Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation 1999; 100: 2473-6.
224. Yokota T, Oritani K, Takahashi I et al.: Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood 2000; 96: 1723-32.
225. Wolf AM, Wolf D, Rumpold H et al.: Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem Biophys Res Commun 2004; 323: 630-5.
226. Fantuzzi G: Adiponectin and inflammation: consensus and controversy. J Allergy Clin Immunol 2008; 121: 326-30.
227. Nawrocki AR, Rajala MW, Tomas E et al.: Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem 2006; 281: 2654-60.
228. Samal B, Sun Y, Stearns G, Xie C et al.: Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol 1994; 14: 1431-7.
229. Moschen AR, Kaser A, Enrich B et al.: Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol 2007; 178: 1748-58.
230. Shin MJ, Hyun YJ, Kim OY et al.: Weight loss effect on inflammation and LDL oxidation in metabolically healthy but obese (MHO) individuals: low inflammation and LDL oxidation in MHO women. Int J Obes (Lond.) 2006; 30: 1529-34.
231. Lynch LA, O´Connell JM, Kwasnik AK et al.: Are natural killer cells protecting the metabolically healthy obese patient? Obesity (Silver Spring) 2009; 17: 601-5.
232. Vaziri ND, Xu ZG, Shahkarami A et al.: Role of AT-1 receptor in regulation of vascular MCP-1, IL-6, PAI-1, MAP kinase, and matrix expressions in obesity. Kidney Int 2005; 68: 2787-93.
233. Stefan N, Kantartzis K, Machann J et al.: Identification and characterization of metabolically benign obesity in humans. Arch Intern Med 2008; 168: 1609-16.
234. Bays HE: „Sick fat,” metabolic disease, and atherosclerosis. Am J Med 2009; 122(1 Suppl): S26-37.
235. Jensen MD: Role of body fat distribution and the metabolic complications of obesity. J Clin Endocrinol Metab 2008; 93(11 Suppl 1): S57-63.
236. Lee CG, Carr MC, Murdoch SJ et al.: Adipokines, inflammation, and visceral adiposity across the menopausal transition: a prospective study. J Clin Endocrinol Metab 2009; 94: 1104-10.
237. Kim JY, van de Wall E, Laplante M et al.: Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 2007; 117: 2621-37.
238. Shibata R, Sato K, Pimentel DR et al.: Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med 2005; 11: 1096-103.
239. Hara K, Boutin P, Mori Y et al.: Genetic variation in the gene encoding adiponectin is associated with an increased risk of type 2 diabetes in the Japanese population. Diabetes 2002; 51: 536-40. Erratum in: Diabetes 2002; 51: 1294.
240. Stumvoll M, Tschritter O, Fritsche A et al.: Association of the T-G polymorphism in adiponectin (exon 2) with obesity and insulin sensitivity: interaction with family history of type 2 diabetes. Diabetes 2002; 51: 37-41.
241. Yang WS, Yang YC, Chen CL et al.: Adiponectin SNP276 is associated with obesity, the metabolic syndrome, and diabetes in the elderly. Am J Clin Nutr 2007; 86: 509-13.
242. Menzaghi C, Ercolino T, Di Paola R et al.: A haplotype at the adiponectin locus is associated with obesity and other features of the insulin resistance syndrome. Diabetes 2002; 51: 2306-12.
243. Zhang N, Shi YH, Hao CF et al.: Association of +45G15G(T/G) and +276(G/T) polymorphisms in the ADIPOQ gene with polycystic ovary syndrome among Han Chinese women. Eur J Endocrinol 2008; 158: 255-60.
244. Fumeron F, Aubert R, Siddiq A et al.: Adiponectin gene polymorphisms and adiponectin levels are independently associated with the development of hyperglycemia during a 3-year period: the epidemiologic data on the insulin resistance syndrome prospective study. Diabetes 2004; 53: 1150-7.
245. Zacharova J, Chiasson JL, Laakso M; STOP-NIDDM Study Group: The common polymorphisms (single nucleotide polymorphism [SNP] +45 and SNP +276) of the adiponectin gene predict the conversion from impaired glucose tolerance to type 2 diabetes: the STOP-NIDDM trial. Diabetes 2005; 54: 893-9.
246. Filippi E, Sentinelli F, Trischitta V et al.: Association of the human adiponectin gene and insulin resistance. Eur J Hum Genet 2004; 12: 199-205.
247. Vozarova de Courten B, Hanson RL et al.: Common Polymorphisms in the Adiponectin Gene ACDC Are Not Associated With Diabetes in Pima Indians. Diabetes 2005; 54: 284-9.
278. Bouatia-Naji N, Meyre D, Lobbens S et al.: ACDC/adiponectin polymorphisms are associated with severe childhood and adult obesity. Diabetes 2006; 55: 545-50.
249. Gu HF, Abulaiti A, Ostenson CG et al.: Single nucleotide polymorphisms in the proximal promoter region of the adiponectin (APM1) gene are associated with type 2 diabetes in Swedish caucasians. Diabetes 2004; 53, Suppl 1: S31-5.
250. Populaire C, Mori Y, Dina C et al.: Does the -11377 promoter variant of APM1 gene contribute to the genetic risk for Type 2 diabetes mellitus in Japanese families? Diabetologia 2003; 46: 443-5.
251. Norata GD, Ongari M, Garlaschelli K et al.: Effect of the -420C/G variant of the resistin gene promoter on metabolic syndrome, obesity, myocardial infarction and kidney dysfunction. J Intern Med 2007; 262: 104-12.
252. Osawa H, Tabara Y, Kawamoto R et al.: Plasma resistin, associated with single nucleotide polymorphism -420, is correlated with insulin resistance, lower HDL cholesterol, and high-sensitivity C-reactive protein in the Japanese general population. Diabetes Care 2007; 30: 1501-6.
253. Mattevi VS, Zembrzuski VM, Hutz MH: A resistin gene polymorphism is associated with body mass index in women. Hum Genet 2004; 115: 208-12.
254. Bouchard L, Weisnagel SJ, Engert JC et al.: Human resistin gene polymorphism is associated with visceral obesity and fasting and oral glucose stimulated C-peptide in the Québec Family Study. J Endocrinol Invest 2004; 27: 1003-9.
255. Cho YM, Youn BS, Chung SS et al.: Common genetic polymorphisms in the promoter of resistin gene are major determinants of plasma resistin concentrations in humans. Diabetologia 2004; 47: 559-65. Epub 2004 Jan 23. Erratum in: Diabetologia 2004; 47: 1337-8.
256. Beckers S, Peeters AV, Freitas F et al.: Analysis of genetic variations in the resistin gene shows no associations with obesity in women. Obesity (Silver Spring) 2008; 16: 905-7.
otrzymano/received: 0000-00-00 zaakceptowano/accepted: 0000-00-00 Adres/address: Pełna wersja artykułu Ocena zależności wydzielania adipokin i insulinooporności oraz asocjacji wybranych polimorfizmów genów adiponektyny i rezystyny w otyłości dostępna w Czytelni Medycznej Borgis. |
  Proszę kliknąć w wybraną okładkę aby przejść na stronę czasopisma
 |






Chcesz być na bieżąco? Polub nas na Facebooku: strona Wydawnictwa na Facebooku |