|
© Borgis - Postępy Nauk Medycznych 9, s. 646-650
*Bartłomiej Noszczyk
Medycyna regeneracyjna w leczeniu Epidermolysis bullosa – rola chirurgii plastycznej
Regenerative medicine in the treatment for Epidermolysis bullosa – the role of plastic surgery
Klinika Chirurgii Plastycznej Centrum Medycznego Kształcenia Podyplomowego w Warszawie
Kierownik Kliniki: prof. dr hab. med. Józef Jethon Streszczenie
Epidermolysis bullosa jest grupą rzadkich wrodzonych chorób skóry i nabłonków, w których defekty genetyczne powodują uszkodzenia białek okolicy błon podstawnych. Leczenie jest objawowe, a jego podstawę stanowią interwencje chirurgiczne, mogące jedynie poprawiać stan bliznowatych przykurczów rąk i stóp, palcozrostów, zwężeń przełyku, ran i nowotworów skóry. Częste nawroty zmian są nieuniknione. Nadzieję na skuteczniejsze leczenie przyniósł rozwój terapii genowej, a w ostatnich latach medycyna regeneracyjna z terapią komórkową. Dyscypliny te wpłyną wkrótce na kształt chirurgii rekonstrukcyjnej, dając jej do dyspozycji narzędzia skuteczniejsze niż tradycyjne interwencje chirurgiczne jak płodowe przeszczepy szpiku, lub przeszczepy komórek autogennych o wywoływanej pluripotencjalności. Artykuł jest krótkim przeglądem najbardziej interesujących koncepcji badawczych, które mają szansę trafić wkrótce do praktyki klinicznej. Słowa kluczowe: epidermolysis bullosa, medycyna regeneracyjna, chirurgia ręki, palcozrosty
Summary
Epidermolysis bullosa compromise a group of rare diseases of skin and epithelia, where genetic mutations results in synthesis of defected proteins responsible for basement membrane zone integrity. Treatment is symptomatic only, and based on surgical interventions which ameliorate conditions of contractured hands and feet, syndactyly, esophageal stenosis, wounds and skin tumors. Frequent recurrences are unavoidable. Some prospects for more effective treatment modalities brought gene therapy development, and recently regenerative medicine with cellular therapy. These disciplines influence reconstructive surgery, by measures more effective than traditional surgical interventions, like embrional bone marrow transplantation or induction of autogenous pluripotent stem cells. This paper is a review of some most interesting scientific concepts, which may soon become a part of clinical practice. Key words: epidermolysis bullosa, regenerative medicine, hand surgery, syndactyly
Leczenie chirurgiczne
Wrodzone postacie pęcherzowego spływania naskórka ( Epidermolysis bullosa – EB) są stosunkowo rzadkimi chorobami nabłonków, rozpoznawanymi i początkowo leczonymi w oddziałach dermatologicznych. W wielu krajach pacjenci kierowani są następnie do wyspecjalizowanych szpitali, gdzie dalsze leczenie prowadzone jest przez lekarzy różnych specjalności. Tak jest między innymi w Polsce, gdzie dzieci z najcięższymi postaciami EB trafiają do Szpitala Dziecięcego im. Prof. Bogdanowicza w Warszawie. Wśród lekarzy zaangażowanych w leczenie EB są tu: chirurdzy, anestezjolodzy, pediatrzy, okuliści, stomatolodzy, ortopedzi, rehabilitanci oraz psycholodzy. Ośrodek współpracuje dodatkowo z oddziałami dermatologii oraz genetyki. Wśród specjalistów znajduje się również chirurg plastyk, który podobnie jak w bliźniaczych ośrodkach w innych krajach, służy pomocą jako konsultant i operator.
Epidermolysis bullosa należy do grupy wrodzonych chorób nabłonków i skóry, w których różne mutacje w obrębie genów kodujących białka okolicy błon podstawnych odpowiadają za ich podatność, i skłonność do tworzenia pęcherzy i ran. Ciągle powtarzające się rany o różnej zdolności do gojenia prowadzą do powikłań w postaci owrzodzeń, przykurczów i nowotworów. Najbardziej narażona jest powierzchnia skóry, przy czym niemal równie często powikłania dotyczą jamy ustnej i przełyku. Wśród dwóch najcięższych form EB znajduje się postać dystroficzna (DEB) charakteryzująca się powstawaniem narastających przykurczów w obrębie rąk i stóp oraz postać łącząca (graniczna) (JEB), prowadząca w niektórych formach do zagrażających życiu ran na olbrzymiej powierzchni ciała (1).
W postaci dystroficznej chorzy rodzą się z zaburzoną zdolnością do produkcji kolagenu typu VII, który odpowiada za tworzenie włókien kotwiczących błony podstawne do zrębu. W naskórku przejawia się to powstawaniem pęcherzy oddzielających się wraz z błoną podstawną, co w efekcie odsłania skórę właściwą i prowadzi do gojenia z powstawaniem blizn. Z czasem tworzą się przykurcze rąk i stóp zwijające niekiedy kończynę w kokon pokryty rogowaciejącym naskórkiem (ryc. 1). W postaci łączącej, defekt dotyczy białka powierzchni błony podstawnej, to jest lamininy-5, lub białek przezbłonowych jak kolagen XVII, lub integryna alfa6beta4 (2). W najcięższej formie JEB uszkodzenie różnych genów kodujących łańcuchy lamininy prowadzi do ran, które w okresie niemowlęcym stwarzają poważne zagrożenie życia (ryc. 2).
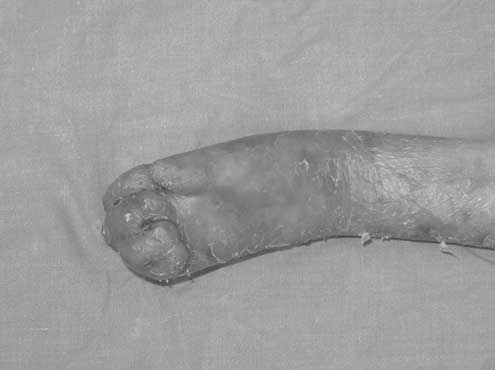 Ryc. 1. Silnie zwinięta w kokon ręka w dystroficznej postaci Epidermolysis bullosa (DEB).
 Ryc. 2. Plecy dziecka z graniczną postacią Epidermolysis bullosa (JEB). Zmiany w tej postaci są często letalne.
Leczenie chirurgiczne w DEB polega na uwalnianiu przykurczów i opatrunkach prowadzonych do pełnego zagojenia. Ponieważ choroba ma charakter genetyczny, nawrót przykurczów jest nieunikniony, następując średnio po 1,5-2 lat. Wykonywanie kolejnych operacji ma jednak sens, ułatwiając prawidłowy psychofizyczny rozwój dziecka. Podstawowym problemem napotykanym podczas operacji jest ogromna podatność naskórka na urazy. Nawet ułożenie dziecka na stole chirurgicznym wymaga w takich warunkach doświadczenia. Wykluczone jest stosowanie jakichkolwiek przylepców na skórę, a znieczulenie powinno być prowadzone bez intubacji w celu uniknięcia uszkodzeń w jamie ustnej i górnych drogach oddechowych.
Rękę operuje się zazwyczaj bez niedokrwienia, co z kolei pozwala uniknąć okrężnych ran na ramieniu. Przecięcie zrogowaciałego naskórka wiąże się z dostaniem się krwi pod jego powierzchnię. Zarówno to, jak i dalsze manipulacje oddzielają naskórek od skóry właściwej daleko poza planowanym polem operacyjnym. Stąd operację lepiej jest zaczynać od zdjęcia naskórka z całej ręki, w manewrze określanym jako „zdjęcie rękawiczki”. Zwiększa to powierzchnię pola operacyjnego, ale ogranicza dalsze uszkodzenia do granicy nadgarstka (3).
Uwolnienie przykurczów i wyprostowanie palców wiąże się często z odsłonięciem pęczków naczyniowo-nerwowych i pochewek ścięgien zginaczy. Tak głębokie ubytki pokrywa się czasami autogennymi przeszczepami skóry pełnej grubości (4). Jednak pobieranie i przygotowywanie przeszczepów prowadzi zazwyczaj do oderwania warstwy naskórka, zarówno na przeszczepie, jak i w miejscu pobrania. W takiej sytuacji do wszycia pozostaje jedynie warstwa skóry właściwej, co wprawdzie nie pogarsza wgajania, ale przysparza ran. Podobne problemy związane są z wykorzystywaniem dermatomowych przeszczepów niepełnej grubości. Zastosowanie dermatomu zrywa stycznie naskórek na dużej powierzchni. Przeszczepiane warstewki skóry właściwej nie mają dużej wartości, a pozostawione rany mogą goić się w trudny do przewidzenia sposób. Wielu autorów zatem rezygnuje ze stosowania przeszczepów w ogóle (5). Liczne obserwacje świadczą, że gojenie może być niezależne od wykorzystania przeszczepów. Trwa jednak różnie długo, według niektórych zajmując zaledwie miesiąc, przy czym częściej przedłużając się do 5-8 tygodni. W tym czasie cotygodniowe opatrunki wymagają wykorzystania bloku operacyjnego i znieczulenia ogólnego. Zwiększa to bardzo koszty leczenia, narażając przy tym dziecko na olbrzymi stres. Ponieważ nawroty w operowanej ręce pojawiają się już po 2 latach, a drugą rękę operuje się zwykle w niecały rok po pierwszej, to kontakt pacjenta ze szpitalem jest praktycznie stały. Współczesna nauka oferuje już jednak rozwiązania, które w najbliższych latach odmienią prawdopodobnie los wielu dzieci z EB. Należy do nich terapia genowa (1), oraz wykorzystywana już niekiedy w praktyce terapia komórkowa. Oba kierunki badań należą do nowej dyscypliny naukowej określanej coraz częściej mianem medycyny regeneracyjnej.
Medycyna regeneracyjna
Preparaty sztucznej skóry oraz hodowle tkankowe projektowane do stosowania w klinice, powstają w laboratoriach inżynierii tkankowej, z których wiele współpracuje lub należy do zespołów tworzonych przez chirurgów plastycznych (6). Dostępne obecnie na rynku produkty inżynierii tkankowej ograniczają się wprawdzie jedynie do preparatów sztucznej skóry, jednak wkrótce wybór poszerzony zostanie o preparaty nadające się do odtwarzania tkanki łącznej, kości, chrząstki, tkanki tłuszczowej, naczyń krwionośnych i piersi. Przez swą naturalną interdyscyplinarność chirurgia plastyczna i rekonstrukcyjna już dziś bierze udział w planowaniu tych badań (7). W leczeniu Epidermolysis bullosa rola plastyka wydaje się być szczególna, polegając zarówno na wypełnianiu zadań chirurga, jak i znajomości współczesnych badań w medycynie regeneracyjnej (8). Wprowadzanie ich wyników do zastosowań klinicznych wydaje się mieć wówczas szczególnie mocny grunt, niosąc uzasadnione nadzieje na przyszłość.
W leczeniu DEB wyniki zaczynają być już widoczne. Niedawno zauważono, że kolagen typu VII podawany śródskórnie w przeszczepioną na grzbiet myszy skórę chorych z DEB, w sposób wyraźny poprawia jej stan (9).
Z podobnych metod korzystają często chirurdzy plastycy, którzy wykorzystują kolagen w celach kosmetycznych, podając niewielkie objętości w śródskórnych zastrzykach, wypełniając zmarszczki na twarzy. W celach estetycznych podawany jest jednak wołowy kolagen typu I, który nie ma zastosowania w pęcherzowym spływaniu naskórka. W przypadku wymienionej pracy wykorzystano rekombinowany kolagen VII syntezowany przez ludzkie komórki in vitro. Ostatnio ta sama grupa badaczy zastosowała opracowany przez siebie sposób produkcji białka do miejscowego leczenia skóry w mysim modelu DEB (10). Poprawiało to znacznie stan skóry zwierząt. Co ciekawe, włókna białka ulegały samoistnej organizacji we włókna kotwiczące, zachowując się „inteligentnie”. Przyczyny tego zjawiska nie są znane, niemniej wróży ono możliwość zastosowań kolagenu typu VII w leczeniu chorych. Problem polega jednak na tym, że synteza kolagenu VII opracowana została dla celów doświadczalnych, nie mając zastosowań technologicznych. Ilość ludzkiego kolagenu potrzebna do leczenia jednego pacjenta wielokrotnie przekracza możliwości opisanej metody (11).
Kilka lat temu doniesiono, że fibroblasty z wprowadzonym genem kodującym produkcją kolagenu VII, podejmują syntezę białka, zarówno w skórze myszy, jak i skórze ludzkiej przeszczepionej na grzbiet myszy (12). Podstawowe znaczenie tego doświadczenia polegało na wykorzystaniu fibroblastów, które w zdrowej skórze nie produkują wprawdzie kolagenu typu VII, jednak poprzez łatwość hodowli i małą immunogenność, nadają się znakomicie do zastosowań klinicznych. Dzięki spostrzeżeniu zauważono, że śródskórne wstrzyknięcia kontrolnych, nie zmienionych genetycznie komórek, prowadziły również do zwiększonej syntezy białka. Było to bezpośrednią przyczyną pierwszych badań klinicznych, które mogą zadecydować o najbliższym kształcie terapii w EB. W badaniu chorym z DEB wykonywano jednorazowe śródskórne zastrzyki niewielkiej liczby allogenicznych fibroblastów pochodzących od nieskojarzonych dawców, lub od rodziców, którzy są heterozygotycznymi nosicielami jednej kopii zmutowanego genu (13). Prowadziło to do długotrwałego zwiększenia produkcji kolagenu typu VII w miejscu podania komórek. Co więcej, działanie fibroblastów powodowało kliniczną poprawę skóry w miejscu zastrzyku, przejawiającą się mniejszą podatnością na urazy oraz poprawą histologiczną w postaci zwiększonej ilości włókien kotwiczących.
Okazuje się jednak, że pojawiający się w skórze kolagen VII produkowany był nie przez podane allogeniczne fibroblasty, lecz przez keratynocyty chorego. Zwiększona produkcja prowadziła zatem do odkładania większej ilości uszkodzonego białka. Fibroblasty wywierały działanie parakrynne na keratynocyty, którego skutki widoczne były jeszcze długo po zniknięciu allogenicznych komórek ze skóry. Mimo nieprawidłowej budowy, produkowane dodatkowe białko ograniczało jednak możliwość tworzenia pęcherzy.
Badanie niesie ze sobą cały szereg pytań, na które trzeba będzie odpowiedzieć zanim nowe leczenie będzie mogło być szerzej stosowane (14). Jedno z nich dotyczy rzeczywistego efektu terapeutycznego po stosowaniu większych dawek komórek. Nie wiadomo jak długo efekt pojedynczej miejscowej dawki może się utrzymywać, ani ile dawek potrzeba by poprawić kliniczny stan skóry choćby w obrębie samych rąk. Nie wiadomo również czy większe dawki allogenicznych komórek nie spowodują reakcji układu odpornościowego gospodarza. Bardzo interesującym zagadnieniem pozostaje również wpływ żywych komórek ludzkich wbudowanych w dostępne na rynku substytuty skóry. W jedynych wykonanych dotychczas badaniach wpływ żywego konstruktu na długość remisji nie był jednoznaczny, wykazując jednak pewne cechy przewagi nad gojeniem bez zastosowania produktu (15). Zastosowanie większej ilości żywych komórek w konstrukcie mogłoby zapewne efekt ten poprawić.
Możliwość miejscowej korekcji zmian skórnych wykazano niedawno również u chorego z JEB, któremu przeszczepiono własny naskórek, zmodyfikowany wektorem retrowirusowym do produkcji lamininy-5. Przygotowane ex vivo przeszczepy naskórka, nadawały się do przeniesienia na rany pacjenta, i po wgojeniu pozostawały wolne od nowych pęcherzy przez przynajmniej jeden rok obserwacji. Badanie histologiczne zagojonej skóry wykazywało, że za poprawę kliniczną odpowiadają rzeczywiście zmodyfikowane terapią genową komórki (16).
Należy jednak pamiętać, że mimo dużej skuteczności, wektory wirusowe w terapii genowej niosą poważne ryzyko działań ubocznych (17). Również skuteczność leczenia miejscowego pozostaje ograniczona w porównaniu do potencjalnych efektów terapii systemowej. Próby leczenia systemowego są już jednak podejmowane, i wydają się nieść nadzieje na przyszłość. Wiadomo między innymi, że komórki szpiku kostnego są mobilizowane do krwi i biorą udział w gojeniu ran o dużej powierzchni (18). Bardzo prawdopodobne zatem, że leczenie ran w EB możliwe będzie na drodze systemowego podawania komórek. Pierwszych dowodów dostarczyło niedawne badanie z wykorzystaniem allogenicznych fibroblastów podawanych dożylnie myszom. Zmodyfikowane genetycznie fibroblasty ludzkie pochodzące od chorych z DEB, wykazywały normalną produkcję kolagenu VII, osadzając się w ranach w mysiej skórze. Po podaniu komórek rany wykazywały przyspieszone gojenie (19).
Niezwykle interesujące wyniki uzyskano również po dożylnym podaniu komórek szpiku, transgenicznym myszom służącym jako model dla leczenia DEB (20). Myszy z uszkodzonym genem ColVIIa1nie produkują kolagenu w ogóle, zdychając po kilku dniach po urodzeniu. Autorzy wykazali jednak, że podanie wybranych komórek allogenicznego szpiku innych myszy może przedłużyć życie i znacznie poprawić stan skóry. W doświadczeniu wykorzystali szpik kostny pobrany od innych transgenicznych myszy z wszczepionym genem odpowiedzialnym za produkcję zielonego białka fluorescencyjnego (GFP). Wszystkie komórki tych myszy świecą w świetle ultrafioletowym na zielono. Pobrany szpik GFP podano chorym zwierzętom, wstrzykując stosunkowo dużą ilość komórek do żyły twarzowej. Nie przyniosło to żadnych widocznych skutków, podobnie jak nieskuteczne okazało się podawanie innych słabo zróżnicowanych komórek od dorosłych osobników GFP, jak multipotencjalne komórki progenitorowe, mezenchymalne komórki macierzyste, komórki macierzyste naskórka i przejściowo pobudzone komórki naskórka. W następnej kolejności autorzy poddali szpik procesowi selekcji, wzbogacając wybraną frakcję o komórki wykazujące ekspresję cząsteczek sygnalizujących aktywację limfocytów SLAM (CD150+ CD48–). Frakcja ta zawiera zwiększoną ilość komórek prekursorowych ulegających szybkiej repopulacji po przeszczepach szpiku. Tym razem dożylny wlew dużej ilości wybranych komórek przyniósł efekt w postaci przedłużenia średniego czasu życia zwierząt. Jednak zaledwie u 15% osobników stan skóry poprawił się na tyle, by życie przedłużyć do normalnego okresu. To zaledwie częściowe powodzenie autorzy tłumaczą brakiem wcześniejszej mieloablacji u biorców oraz faktem, że komórki GFP z prawidłową kopią ColVIIa1, wgajały się głównie w nabłonkach przewodu pokarmowego, gdzie zwierzęta miały najwięcej ran. Wyczerpywało to szybko pulę komórek dostępnych, ograniczając działanie na skórę.
Podobnie przełomowe znaczenie może mieć praca badająca możliwości leczenia EB za pomocą przeszczepiania komórek szpiku płodom (21). Zarówno w cytowanej wyżej, jak i obecnej pracy autorzy podjęli próbę przeszczepiania komórek szpiku kostnego do krwi chorych zwierząt. W obu wypadkach zatem nie rozważano zagadnień pełnego przeszczepu szpiku, to jest leczenia podejmowanego np. w białaczkach, i mogącego wymagać mieloablacji. Mieloablacja, czyli zniszczenie własnego szpiku u biorcy przeszczepu, wykonywana jest głównie z powodu problemów z dopasowaniem antygenów zgodności tkankowej HLA w komórkach dawcy. Przy braku dopasowania mieloablacja zapobiega chorobie „przeszczep przeciw gospodarzowi”. W pierwszym trymestrze ciąży u ludzi, płód akceptuje antygeny allogeniczne, wytwarzając tolerancję immunologiczną poprzez niszczenie allogenicznych limfocytów w grasicy. W tym okresie prawdopodobnie możliwe byłoby przeszczepianie komórek szpiku bez dopasowania immunologicznego, i bez ryzyka wytworzenia odporności na białka produkowane przez przeszczepione komórki. Pierwsze próby u ludzi będą już wkrótce wykonywane (22). W przypadku leczenia EB przeszczep komórek szpiku do krwi chorego płodu (E-BMT) może mieć dodatkową zaletę wytwarzania tolerancji dla prawidłowego kolagenu typu VII. Istnieje bowiem przypuszczenie, że prawidłowe białko podawane lub wytworzone w wyniku leczenia w organizmach chorych dzieci, może być rozpoznawane jako obce i konsekwentnie niszczone. Autorzy wykonywali przeszczepy E-BMT płodom myszy z wrodzonym EB. Przedłużało to okres życia chorych zwierząt z dwóch do 17-19 dni. Jednak żadne zwierze nie przeżyło powyżej 3 tygodni. Leczenie prowadziło do pojawienia się kolagenu VII w skórze i osłabienia objawów spływania naskórka. Świadczyło to o pojawieniu się tolerancji dla allogenicznych komórek i produkowanego przez nie prawidłowego białka. Mimo że nie udało się całkowicie zapobiec objawom choroby, autorzy są zdania, że przeszczepy E-BMT w leczeniu Epidermolysis powinny być rozwijane, gdyż mogą się wkrótce stać alternatywą dla obecnych, mało skutecznych metod leczenia (21).
Największe nadzieje wiązać jednak można z zupełnie innym kierunkiem badań. Poszukiwanie i badanie komórek macierzystych dorosłych organizmów może umożliwić postęp w wielu dziedzinach medycyny odtwórczej, na długo jednak ograniczone pozostanie barierą immunologiczną. Badania komórek embrionalnych będą z kolei ograniczone barierą zagadnień etycznych. Można jednak przewidywać, że wkrótce możliwe będzie cofanie zróżnicowania dowolnych komórek z dojrzałych tkanek do stadium komórek pluripotencjalnych. Potwierdzono to już w 2006 roku, wprowadzając do mysich komórek skóry 4 geny z pomocą wektora wirusowego (23). Spowodowało to przeprogramowanie ich stanu do etapu wczesnych komórek płodowych. Ten wywołany stan pluripotencjalności (induction of pluripotent stem cells – IPS) nadaje im zdolność rozwoju zarodka, a więc i potencjalną możliwość kierowania ich rozwojem dla potrzeb medycyny regeneracyjnej. Wykorzystanie wektorów wirusowych dyskwalifikuje jednak to osiągnięcie z zastosowań klinicznych. Ostatnio inna grupa badaczy udowodniła, że przeprogramowanie ludzkich i mysich komórek somatycznych genami c-Myc, Klf4, Oct4i Sox2jest możliwe bez stosowania wirusów. Autorzy wykorzystali transpozon piggyBAC z systemem transpozazy, którego zaleta polega na insercjach genów w określonych miejscach i możliwość ich kontrolowanego usuwania (24). Autorzy wprawdzie przyznają, że system wymaga nadal usprawniania, jednak dostrzegają również jego zastosowanie w medycynie regeneracyjnej. Rzeczywiście, obecnie można sobie łatwo wyobrazić, że odkrycie pozwoli z pobranych komórek autogennych wytworzyć nowe narządy, z których skóra wydaje się być najłatwiejsza. Ten sam system wykorzystać można do wymiany zmutowanych genów kolagenu VII lub lamininy-5.
Piśmiennictwo
1. Noszczyk B, Jethon J: Terapia genowa w pęcherzowym spływaniu naskórka ( Epidermolysis bullosa). Polski Przegląd Chirurgiczny 2008; 80(4): 389-397.
2. Fine JD et al.: Revised classification system for inherited Epidermolysis bullosa: Report of the Second International Consensus Meeting on diagnosis and classification of Epidermolysis bullosa. J Am Acad Dermatol 2000; 42(6): 1051-66.
3. Noszczyk B, Jethon J: Leczenie chirurgiczne w Epidermolysis bullosa. Polski Przegląd Chirurgiczny 2008; 80(5): 485-496.
4. Ladd AL, Kibele A, Gibbons S: Surgical treatment and postoperative splinting of recessive dystrophic Epidermolysis bullosa. J Hand Surg [Am] 1996; 21(5): 888-97.
5. Noszczyk B et al.: Wyniki operacyjnego i poooperacyjnego leczenia palcozrostów w Epidermolysis bullosa. Polski Przegląd Chirurgiczny 2008; 80(8): 744-759.
6. Bannasch H et al.: Tissue engineering of skin substitutes. Panminerva Med 2005; 47(1): 53-60.
7. Sterodimas A et al.: Tissue engineering in plastic surgery: an up-to-date review of the current literature. Ann Plast Surg 2009; 62(1): 97-103.
8. Caterson EJ, Caterson SA: Regeneration in medicine: a plastic surgeons „tail” of disease, stem cells, and a possible future. Birth Defects Res C Embryo Today 2008; 84(4): 322-34.
9. Woodley DT et al.: Intradermal injection of lentiviral vectors corrects regenerated human dystrophic Epidermolysis bullosa skin tissue in vivo. Mol Ther 2004; 10(2): 318-26.
10. Remington J et al.: Injection of recombinant human type VII collagen corrects the disease phenotype in a murine model of dystrophic Epidermolysis bullosa. Mol Ther 2009; 17(1): 26-33.
11. Bruckner-Tuderman L: Can type VII collagen injections cure dystrophic Epidermolysis bullosa? Mol Ther 2009; 17(1): 6-7.
12. Woodley DT et al.: Normal and gene-corrected dystrophic Epidermolysis bullosa fibroblasts alone can produce type VII collagen at the basement membrane zone. J Invest Dermatol 2003; 121(5): 1021-8.
13. Wong T et al.: Potential of fibroblast cell therapy for recessive dystrophic Epidermolysis bullosa. J Invest Dermatol 2008; 128(9): 2179-89.
14. Poocheron V, Hu S, Kirsner RS: Allogeneic cell therapy for Epidermolysis bullosa. J Invest Dermatol 2008; 128(9): 2134.
15. Fivenson DP, Scherschun L, Cohen LV: Apligraf in the treatment of severe mitten deformity associated with recessive dystrophic Epidermolysis bullosa. Plast Reconstr Surg 2003; 112(2): 584-8.
16. Mavilio F et al.: Correction of junctional Epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med 2006; 12(12): 1397-402.
17. Nielsen TT et al: Incorporating double copies of a chromatin insulator into lentiviral vectors results in less viral integrants. BMC Biotechnol 2009; 9: 13.
18. Rea S et al.: Bone marrow-derived cells in the healing burn wound-More than just inflammation. Burns 2008; 24.
19. Woodley DT et al.: Intravenously injected human fibroblasts home to skin wounds, deliver type VII collagen, and promote wound healing. Mol Ther 2007; 15(3): 628-35.
20. Tolar J et al.: Amelioration of epidermolysis bullosa by transfer of wild-type bone marrow cells. Blood 2009; 113(5): 1167-74.
21. Chino T et al.: Bone marrow cell transfer into fetal circulation can ameliorate genetic skin diseases by providing fibroblasts to the skin and inducing immune tolerance. Am J Pathol 2008; 173(3): 803-14.
22. Troeger C et al.: In utero haematopoietic stem cell transplantation. Experiences in mice, sheep and humans. Swiss Med Wkly 2006; 136(31-32): 498-503.
23. Takahashi K, Yamanaka S: Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126(4): 663-76.
24. Woltjen K et al.: piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009; 1.
otrzymano/received: 2009-05-21 zaakceptowano/accepted: 2009-08-12 Adres/address: *Bartłomiej Noszczyk Klinika Chirurgii Plastycznej Centrum Medycznego Kształcenia Podyplomowego ul. Czerniakowska 231, 00-416 Warszawa tel.: (0-22) 629-69-69 e-mail: noszczyk@melilot.pl Pełna wersja artykułu Medycyna regeneracyjna w leczeniu Epidermolysis bullosa – rola chirurgii plastycznej dostępna w Czytelni Medycznej Borgis. |
  Proszę kliknąć w wybraną okładkę aby przejść na stronę czasopisma
 |






Chcesz być na bieżąco? Polub nas na Facebooku: strona Wydawnictwa na Facebooku |