|
© Borgis - Postępy Nauk Medycznych 8, s. 501-503
*Tadeusz Dębniak, Jan Lubiński
Genetyka kliniczna czerniaka
Clinical genetics of malignant melanoma
Międzynarodowe Centrum Nowotworów Dziedzicznych Zakład Genetyki i Patomorfologii Pomorska Akademia Medyczna, Szczecin
Kierownik Zakładu: prof. dr hab. med. Jan Lubiński Streszczenie
Czerniak złośliwy jest jednym z najbardziej agresywnych nowotworów, jego częstość wzrasta gwałtownie w ostatnich latach. Zwiększone ryzyko zachorowania na czerniaka u potomstwa osób chorych na ten nowotwór jak również rodzinne agregacje tego nowotworu sugerują, że predyspozycja genetyczna jest istotnym czynnikiem uczestniczącym w patogenezie czerniaka. Rodzinny czerniak stanowi najprawdopodobniej heterogenną grupę przypadków o różnym typie dziedziczenia, w większości przypadków wielogenowym. Nierzadko obserwuje się jednak rodzinne agregacje wykazujące cechy autosomalnie dominującego typu dziedziczenia, charakterystycznego dla chorób jednogenowych o wysokiej penetracji. Podłoże genetyczne czerniaka jest złożone i zależne od wielu genów. Głównym genem ryzyka jest CDKN2A. Częsta konstytucyjna zmiana tego genu – A148T, zwiększa ryzyko zachorowania na czerniaka niezależnie od nowotworowego wywiadu rodzinnego. Mutacje genów ARF oraz CDK4, związane z wysokim ryzykiem zachorowania na MM, są niezwykle rzadkie i nie mają istotnego znaczenia w praktyce klinicznej. W większości rodzinnych czerniaków mutacje genu CDKN2A nie występują, co wskazuje na potrzebę identyfikacji nowych genów związanych z predyspozycją do tego nowotworu. Poznano kilka genów/mutacji umiarkowanie modyfikujących ryzyko MM. Ich lista obejmuje XPD, MC1R, BRCA2. Wdrożenie odpowiednich programów diagnostyczno- -profilaktycznych oraz leczniczych może zmniejszyć zachorowalność i śmiertelność. Testy genetyczne oraz analizy danych rodowodowo-klinicznych powinny być wykonywane u wszystkich pacjentów z rozpoznanym czerniakiem, także w przypadkach z ujemnym wywiadem rodzinnym. Słowa kluczowe: czerniak (MM), CKDN2
Summary
Malignant melanoma (MM) represents one of the most aggressive neoplasms and its frequency is increasing rapidly. Increased melanoma risk among relatives of MM patients and familial aggregations of this malignancy point at genetic predisposition as an important factor of MM pathogenesis. Familial MM constitutes most probably a heterogenous group of disorders characterized by occurrence of MM among relatives. The mode of inheritance is controversial and most likely polygenic, however not infrequently, within large families aggregations of MM is consistent with autosomal dominant inheritance. The genetic basis of MM is complex and appears to involve multiple genes. CDKN2A is regarded as the major MM susceptibility gene. In the Polish population common CDKN2A variant (A148T) increases significantly melanoma risk regardless of the cancer family history. Mutations of other high risk genes, ARF and CDK4 are extremely rare and thus clinically insignificant. In majority of MM cases CDKN2A mutations are not found. It is thus necessary to perform association studies focused on identifying genetic markers that could be used in identifying patients with a high risk of MM. List of other genes that carry mutations, which are believed to be associated with moderate MM risk include XPD, MC1R, BRCA2. The management with individuals being at increased MM risk involves clinical screening according to carefully planned surveillance schedule and early treatment of MM tumour. The appropriate management may reduce morbidity and mortality. Genetic testing and clinical evaluation should be performed, and family history should be obtained in all patients affected with MM, also in those with apparently sporadic tumours. Key words: malignant melanoma (MM), CDKN2
Czerniak złośliwy (MM) jest jednym z najbardziej agresywnych nowotworów złośliwych. Każdego roku w Polsce jest odnotowywanych niemal 2 tys. nowych zachorowań na czerniaka (1).
Liczba zachorowań na ten nowotwór wśród ludzi rasy białej dramatycznie wzrosła w ostatnich latach – niemal 10-krotnie w ciągu ostatnich 50 lat (2). Uważa się, że jednym z głównych czynników sprawczych wystąpienia czerniaka jest promieniowanie ultrafioletowe (3). Szczególnie niebezpieczne wydają się być oparzenia słoneczne w dzieciństwie (4). Kolejnymi czynnikami ryzyka są: 1) dysplastyczne znamiona barwnikowe; 2) duża liczba (>100) znamion barwnikowych; 3) jasna karnacja skóry (typ I i II skóry) (5, 6).
Zwiększone ryzyko zachorowania na czerniaka u potomstwa osób chorych na ten nowotwór (7, 8) jak również rodzinne agregacje tego nowotworu sugerują, że predyspozycja genetyczna jest kolejnym istotnym czynnikiem uczestniczącym w patogenezie czerniaka. Rodzinną agregację czerniaka można zdefiniować jako: 1) wystąpienie czerniaka złośliwego u przynajmniej dwóch krewnych I°; lub 2) wystąpienie czerniaka u przynajmniej dwóch krewnych I° lub II°. Stwierdza się ją w około 3- -15% wszystkich zdiagnozowanych przypadków czerniaka (9). W naszym ośrodku wśród 665 nieselekcjonowanych pacjentów rodzinna agregacja u krewnych I° występowała w 24 przypadkach (3,6%).
W części rodzin opisano współistnienie czerniaka skóry oraz gałki ocznej (10). Jak dotąd nie wiadomo jednak czy czerniak gałki ocznej jest częścią zespołu rodzinnego czerniaka skóry (ryc. 1).
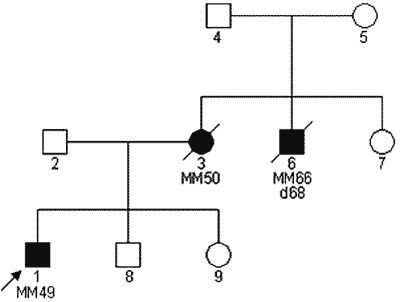 Ryc. 1. Rodzinne występowanie czerniaka u krewnych I° probanta z rozpoznanym czerniakiem złośliwym.
W części rodzin występuje zwiększone ryzyko MM oraz nowotworów złośliwych innych narządów, takich jak rak trzustki, piersi czy guzy ośrodkowego układu nerwowego (11, 12, 13, 14). Podjęte w naszym Ośrodku badania rodzin z silną rodzinną agregacją nowotworów różnych narządów (CFA) sugerują trzykrotnie zwiększone ryzyko wystąpienia raka piersi w młodym wieku (przed 50 r.ż.) wśród krewnych I° pacjentów z MM rozpoznanym do 55. roku życia.
Rodzinny czerniak stanowi najprawdopodobniej heterogenną grupę przypadków o różnym typie dziedziczenia, w większości przypadków wielogenowym (15). Nierzadko obserwuje się jednak rodzinne agregacje wykazujące cechy autosomalnie dominującego typu dziedziczenia, charakterystycznego dla chorób jednogenowych o wysokiej penetracji.
Czerniak złośliwy spowodowany mutacjami konstytucyjnymi genu CDKN2A
Czerniak złośliwy występuje ze zwiększoną częstością u nosicieli mutacji genu CDKN2A (16, 17). Penetracja tego genu jest zmienna i zależna od wieku jak również położenia geograficznego (18). Mutacje germinalne genu CDKN2A wykryto w 46% rodzinnych czerniaków we Francji, 18% rodzinnych przypadków w Stanach Zjednoczonych, 8% w Szwecji i mniej niż 6% w Polsce (19, 20, 21, 22, 23). Przypuszcza się, że częstość występowania zmian w CDKN2A koreluje z liczbą zachorowań na MM w rodzinie oraz młodym wiekiem (<50 r.ż.) (24). Cechami charakterystycznymi dla MM wywołanego zmianami w genie CDKN2A są: 1) rodzinne występowanie; 2) współistnienie raka trzustki i według części autorów raka piersi wśród krewnych; 3) wieloogniskowość.
Badania DNA w diagnostyce MM
Mutacje genów ARF oraz CDK4, związane z wysokim ryzykiem zachorowania na MM, wykryto jak dotąd jedynie w kilku rodzinach na świecie, nie mają, więc istotnego znaczenia w praktyce klinicznej. Za wyjątkiem genu CDKN2A właściwe geny wysokiego ryzyka MM nie zostały jeszcze zidentyfikowane. W większości rodzinnych czerniaków mutacje genu CDKN2A nie występują, co wskazuje na potrzebę identyfikacji nowych genów związanych z predyspozycją do tego nowotworu. Poznano kilka genów/mutacji umiarkowanie modyfikujących ryzyko MM. Współdziałanie średniego ryzyka mutacji w wielu genach oraz dodatkowo wpływ czynników środowiskowych może znacząco zwiększać ryzyko MM. Wydaje się, że uszkodzenia DNA umiarkowanie modyfikujące ryzyko zachorowania odpowiadają za mało nasilone rodzinne agregacje zachorowań.
Przeprowadzone w naszym Ośrodku badania genu CDKN2A u pacjentów z MM wykazały, że częsty wariant A148T ponad dwukrotnie zwiększa ryzyko czerniaka niezależnie od wywiadu rodzinnego, zwłaszcza w młodym (<50 roku życia) wieku (25). Testy DNA można również wykonywać dla takich genów jak MC1R, XPD czy BRCA2. Poziom zwiększenia ryzyka MM dla mutacji/polimorfizmów tych genów jest umiarkowany, w granicach 1,5-3 razy (26, 27, 28). W chwili obecnej w 72% kolejnych nieselekcjonowanych czerniaków badanych w naszym Ośrodku stwierdza się obecność co najmniej jednej z wyżej wymienionych zmian.
Badania skryningowe w rodzinach z czerniakiem
Wszyscy pacjenci z czerniakiem i ich krewni I° oraz II° co 6 miesięcy powinni zgłaszać się na dokładne badanie dermatologiczne. W przypadku obecności znamion dysplastycznych konsultacje powinny odbywać się co 3 miesiące. Zaleca się usuwanie jedynie tych znamion, które wykazują cechy transformacji nowotworowej (powiększanie się zmian, obecność obwódki zapalnej, krwawienie, świąd itp.) oraz są zlokalizowane w miejscach narażonych na urazy mechaniczne.
U pacjentów ze stwierdzoną mutacją w genie CDKN2A lub z rodzinną agregacją czerniaka lub z agregacją czerniaka i raka trzustki wskazane jest wdrożenie odpowiednich programów profilaktyczno-diagnostycznych dla raka trzustki.
Wykonywanie badań kontrolnych piersi od 35-40 roku życia należy przedstawić jako opcję dla kobiet z rodzin z przynajmniej trzema zachorowaniami na nowotwory złośliwe różnych narządów wśród krewnych I° – w tym na czerniaka złośliwego rozpoznanego poniżej 56. roku życia – nie spełniających rodowodowo-klinicznych kryteriów żadnego ze znanych zespołów wysokiej dziedzicznej predyspozycji do nowotworów (CFA). Piśmiennictwo
1. Zatoński W, Tyczyński J: Cancer in Poland in 2003. The Maria-Skłodowska-Curie Memorial Cancer Center, Department of Epidemiology and cancer Prevention, National Cancer Registry, Warsaw 2004.
2. Weinstock MA: Issues in the epidemiology of melanoma. Hematol Oncol Clin North Am 1998, 12: 681-98.
3. English DR, et al.: Sunlight and cancer. Review. Cancer Causes Control 1997, 8: 271-83.
4. Whiteman DC, Whiteman AC, Green AC: Childhood sun exposure as a risk factor for melanoma- a systematic review of epidemiological studies. Cancer causes control 2001, 12: 69-82.
5. Wachsmuth RC, Harland M, Bishop JA: The atypical-mole syndrome and predisposition to melanoma. N Engl J Med 1998, 339: 348-9.
6. Grange F, et al.: Comparison between familial and nonfamilial melanoma in France. Arch Dermatol 1995, 131: 1154-9.
7. Hemminki K, et al.: The nation-wide Swedish family-cancer database-updated structure and familial rates. Acta Oncol 2001, 40: 772-7.
8. Goldgar DE, et al.: Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J Natl Cancer Inst 1994, 86: 1600-8.
9. Berwick M, Wiggins C: The current epidemiology of cutaneous malignant melanoma. Front Biosci. 2006; 11: 1244-54.
10. de Snoo FA, Bergman W, Gruis NA: Familial melanoma: a complex disorder leading to controversy on DNA testing. Fam Cancer 2003, 2: 109-16.
11. Whelan AJ, Bartsch D, Goodfellow PJ: Brief report: a familial syndrome of pancreatic cancer and melanoma with a mutation in the CDKN2tumor-suppressor gene. New Eng J Med 1995, 33: 975-7.
12. Parker JF, et al.: Pancreatic carcinoma surveillance in patients with familial melanoma. Arch Dermatol 2003, 139: 1019-25.
13. Borg A, et al.: High frequency of multiple melanomas and breast and pancreas carcinomas in CDKN2A mutation-positive melanoma families. J Natl Cancer Inst 2000, 92: 1260-6.
14. Kaufman DK, et al.: A familial syndrome with cutaneous malignant melanoma and cerebral astrocytoma. Neurology 1993, 43: 1728-31.
15. Tsao H: Update on familial cancer syndromes and the skin. J Am Acad Dermatol 2000, 42: 939-71.
16. Hussussian CJ, et al.: Germline p16 mutations in familial melanoma. Nat Genet 1994, 8: 15-21.
17. Kamb A, et al.: Analysis of the p16 gene ( CDKN2)as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet 1994, 8: 22-6.
18. Bishop DT, et al.: Melanoma Genetics Consortium. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst 2002, 94: 894-903.
19. Soufir N, et al.: Prevalence of p16 and CDK4 germline mutations in 48 melanoma-prone families in France. The French Familial Melanoma Study Group. Hum Mol Genet 1998, 7: 941.
20. FitzGerald MG, et al.: Prevalence of germ-line mutations in p16, p19ARF, and CDK4 in familial melanoma: analysis of a clinic-based population. Proc Natl Acad Sci USA 1996, 93: 8541-5.
21. Platz A, Hansson J, Ringborg U: Screening of germline mutations in the CDK4, CDKN2C and TP53 genes in familial melanoma: a clinic-based population study. Int J Cancer 1998, 25: 13-5.
22. Platz A, et al.: Screening of germline mutations in the CDKN2A and CDKN2B genes in Swedish families with hereditary cutaneous melanoma. J Natl Cancer Inst 1997, 89: 697-702.
23. Dębniak T, et al.: Germline mutation and large deletion analysis of the CDKN2A and ARF genes in families with multiple melanoma or an aggregation of malignant melanoma and breast cancer. Int J Cancer 2004, 110: 558-62.
24. Bressac-de-Paillerets B, et al.: Genetic and environmental factors in cutaneous malignant melanoma. Biochimie 2002, 84: 67-74.
25. Dębniak T, et al.: The CDKN2A common variants and their association with melanoma risk: a population-based study. Cancer Res 2005, 65: 835-9.
26. Tomescu D, et al.: Nucleotide excision repair gene XPD polymorphisms and genetic predisposition to melanoma. Carcinogenesis 2001, 22: 403-8.
27. Dębniak T, et al. MC1R common variants, CDKN2A and their association with melanoma and breast cancer risk. Int J Cancer 2006, 119: 2597-602.
28. Scott RJ, et al.: BRCA2mutations in a population-based series of patients with ocular melanoma. Int J Cancer 2002, 102: 188-91.
otrzymano/received: 2008-04-23 zaakceptowano/accepted: 2008-07-04 Adres/address: *Tadeusz Dębniak Międzynarodowe Centrum Nowotworów Dziedzicznych, Zakład Genetyki i Patomorfologii, Pomorska Akademia Medyczna ul. Połabska 4, 70-115 Szczecin tel.: (0-91) 466-15-32 e-mail: debniak@wp.pl Pełna wersja artykułu Genetyka kliniczna czerniaka dostępna w Czytelni Medycznej Borgis. |
  Proszę kliknąć w wybraną okładkę aby przejść na stronę czasopisma
 |






Chcesz być na bieżąco? Polub nas na Facebooku: strona Wydawnictwa na Facebooku |