|
© Borgis - Postępy Nauk Medycznych 7, s. 456-459
*Józef Kładny1, Grzegorz Kurzawski2, Janina Suchy2, Jan Lubiński Zespół Lyncha (HNPCC)
Lynch syndrome
1Klinika Chirurgii Ogólnej i Onkologicznej Pomorska Akademia Medyczna, Szczecin
Kierownik Kliniki: dr hab. med. Józef Kładny 2Międzynarodowe Centrum Nowotworów Dziedzicznych Zakład Genetyki i Patomorfologii Pomorska Akademia Medyczna, Szczecin Kierownik Zakładu: prof. dr hab. med. Jan Lubiński Streszczenie
Praca jest przeglądem doświadczeń własnych oraz danych z piśmiennictwa na temat diagnostyki, profilaktyki i leczenia nowotworów w przebiegu zespołu Lyncha (HNPCC). Unikalną wartość wnoszą odmienne od standardowych kryteria rozpoznawania „HNPCC susp”. Taką sytuację kliniczną według własnych badań autorów należy rozpoznawać wówczas, gdy: 1. u probanta lub któregokolwiek z jego krewnych I° lub II° stwierdza się raka jelita grubego; 2. u chorego z rakiem jelita grubego, spełniającego kryterium 1) lub wśród jego krewnych I° stwierdza się raka z tzw. spektrum HNPCC – raka jelita grubego, trzonu macicy, jelita cienkiego lub dróg moczowych; 3. co najmniej jeden z raków spełniających kryteria 1) lub 2) zdiagnozowany został poniżej 50 r.ż. 4. wykluczono polipowatość rodzinną. Słowa kluczowe: HNPCC, kryteria diagnostyczne, prewencja, leczenie
Summary
Publication is the review based on author´s own experience and literature data concerning diagnosis, prevention and treatment of tumors in families with Lynch Syndrome (HNPCC). The unique value is provided by special, distinct from standard, criteria of diagnosing "HNPCC susp”. According to authors studies such diagnosis can be established when: 1. proband or at least one of his I° or II° relatives is affected by colorectal cancer (CRC); 2. patient with CRC matching criterium 1) or one of his I° relatives is affected by at least one cancer from so called HNPCC spectrum – CRC, cancer of the endometrium or of the small bowel or of the urinary tract; 3. at least one of cancers matching criteria 1) or 2) has been diagnosed under age of 50 years; 4. familial adenomatous polyposis is excluded. Key words: HNPCC, diagnostic criteria, prevention, treatment
Szacuje się, że wysoka genetyczna predyspozycja jest przyczyną 10-20% wszystkich raków jelita grubego (RJG) (1, 2, 3, 4, 5). Do dobrze znanych zespołów dziedzicznej predyspozycji do nowotworów w przebiegu których dochodzi do rozwoju RJG należą dziedziczące się zgodnie z prawami Mendla takie zespoły jak: dziedziczny nie związany z polipowatością rak jelita grubego (HNPCC, zespół Lyncha), polipowatość rodzinna w tym zespół Gardnera, zespoły Zankasa, Turcota, Peutz-Jeghersa i polipowatości młodzieńczej.
Zespół Lyncha (HNPCC)
Opisany przez Lyncha (6) w latach sześćdziesiątych zespół dziedzicznego nie związanego z polipowatością RJG stanowi przynajmniej około 5% wszystkich RJG. Wykazano, że HNPCC powstaje w wyniku mutacji jednego z kilku genów takich jak MSH2, MLH1, MSH6, PMS2. Mutacje w obrębie dwóch pierwszych z nich są najczęstszą przyczyną zespołów Lynch (7, 8, 9, 10). Do charakterystycznych cech klinicznych zespołu Lyncha należą:
– wczesny wiek zachorowania (średnio ok. 45 r.ż.)
– częstsza prawostronna lokalizacja guza
– 2 i więcej przypadków RJG wśród krewnych I°
– wiele synchronicznych i metachronicznych ognisk RJG
– występowanie choroby w kolejnych pokoleniach (transmisja pionowa)
– zwiększona częstość występowania wśród krewnych raków trzonu macicy, jelita cienkiego i dróg moczowych.
Według międzynarodowej grupy ekspertów (International Collaborative Group on HNPCC – ICG-HNPCC) zespół Lyncha można jednoznacznie rozpoznawać wówczas, gdy wykryta zostanie mutacja konstytucyjna w jednym z genów związanych z HNPCC jak np. MSH2 czy MLH1 lub też na podstawie danych rodowodowo-klinicznych, wówczas, gdy spełnione są kryteria przedstawione w tabeli 1 (11, 12).
Tabela 1. Kryteria diagnostyczne HNPCC wg ICG-HNPCC (12).
Wszystkie pozostałe parametry (prawostronna lokalizacja, syn- lub metachroniczne guzy) według tych badaczy powinny być traktowane jako cechy dodatkowe.
*wykluczono występowanie mnogich polipów (polipowatości) jelita grubego, wrodzonego przerostu nabłonka barwnikowego siatkówki, torbieli i kostniaków kości twarzoczaszki oraz desmoidów Na rycinie 1 przedstawiono rodzinę spełniającą kryteria definitywnego HNPCC wg ICG-HNPCC.
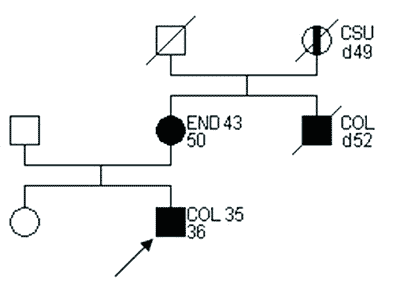 Ryc. 1. Rodowód rodziny z cechami HNPCC wg ICG-HNPCC. (14).
Ze względu na niepełną penetrację genów typową dla chorób mendlowskich dominujących, zgony z powodu różnych chorób, czy też trudności w uzyskaniu pełnych informacji o wszystkich członkach rodzin, znaczna część – być może większość – rodzin w rzeczywistości obciążonych HNPCC, nie może być rozpoznana w oparciu o kryteria wg tabeli 1, tj. amsterdamskie.
Dlatego też szereg autorów proponuje stosowanie innego typu kryteriów, spełnienie których nie upoważnia do jednoznacznego rozpoznania HNPCC, jednak jest pomocne w wykrywaniu rodzin ze zwiększonym ryzykiem (12, 13, 14, 15). Naszym zdaniem w identyfikacji przypadków podejrzanych o HNPCC szczególnie użyteczne są kryteria zestawione w tabeli 2.
Tabela 2. Kryteria rozpoznawania rodzin podejrzanych o HNPCC (16).
Przykłady rodzin spełniających kryteria „podejrzenia HNPCC” przedstawiono na rycinach 2 i 3.
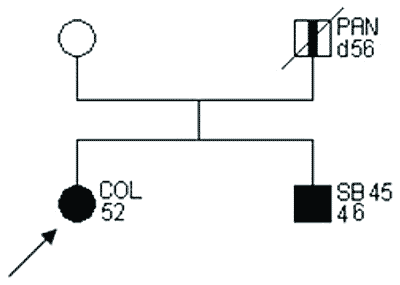 Ryc. 2. Rodowód rodziny „podejrzanej o HNPCC”.
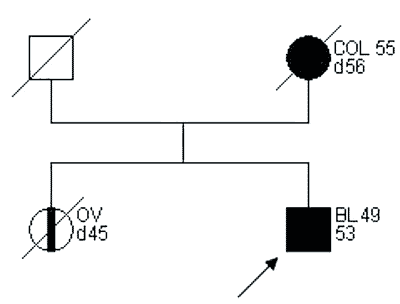 Ryc. 3. Rodowód rodziny „podejrzanej o HNPCC”.
Diagnostyka molekularna nosicielstwa mutacji w genach związanych z rozwojem HNPCC
Wykrycie mutacji markerowej dla rodziny z HNPCC ma bardzo istotne znaczenie kliniczne, ponieważ: A) umożliwia wykluczenie ok. 50% członków rodziny z grupy wysokiego ryzyka. B) ułatwia podjęcie decyzji o radykalności zabiegów chirurgicznych np. kolektomii zamiast klasycznej resekcji odcinkowej oraz profilaktycznej histerektomii i owariektomii w sytuacji, gdy zabiegi te wykonywane są u zdiagnozowanych nosicieli mutacji.
Test MSH2 i MLH1
Wykonanie testów DNA jest wskazane w rodzinach spełniających co najmniej kryteria podejrzenia o HNPCC. Po wykluczeniu FAP (występowanie cech charakterystycznych dla FAP to: polipowatość jelit, przerost nabłonka barwnikowego siatkówki oka, występowanie zmian torbielowato-kostniakowych kości twarzoczaszki i desmoidów), jeżeli dysponujemy tkanką z guza, należy wykonać immunohistochemiczną ocenę ekspresji białek MLH1, MSH2, MSH6w tkance nowotworowej, ponieważ badanie to może zawęzić dalsze postępowanie do poszukiwania mutacji w obrębie jednego genu (brak ekspresji – może wskazywać na zmutowany gen!).
Wieloletnie wieloośrodkowe badania doprowadziły do scharakteryzowania częstości i rodzajów występujących w Polsce mutacji genów MSH2i MLH1 (17). Najważniejsze ustalenia przydatne w opracowaniu testu to:
– najczęstszą przyczyną zespołu Lyncha w Polsce są mutacje w obrębie MSH2i MLH1, które stanowią 90% wszystkich mutacji związanych z tym zespołem;
– mutacje wykrywane testem MLPA stanowią około 10% wszystkich mutacji;
– mutacje powtarzalne występują u ponad 60% wszystkich rodzin z mutacjami.
Biorąc pod uwagę te wytyczne oraz koszty analiz, po immunohistochemii w następnej kolejności należy wykonać badanie MLPA dla MSH2, MLH1. W przypadku wyniku negatywnego następnym krokiem powinno być wykonanie badania najczęstszych charakterystycznych dla populacji polskiej mutacji w MSH2 i MLH1 w DNA z krwi obwodowej pacjenta. Ostatnim etapem wykrywania mutacji jest analiza wszystkich fragmentów kodujących genów za pomocą DHPLC (18) i sekwencjonowanie fragmentów, których chromatogramy wskazują na obecność heterodupleksów.
Prowadzenie rodzin z HNPCC
Posiadana obecnie wiedza na temat tego zespołu wskazuje na konieczność zastosowania specjalnej formy opieki profilaktyczno-diagnostycznej i leczniczej. W poszczególnych ośrodkach obowiązują różne programy (4, 19, 20). W świetle najnowszych danych u członków rodzin z zespołami Lynch uzasadnione wydaje się przyjęcie następujących zasad opieki medycznej:
Optymalizacja diety
U osób z wysokim ryzykiem rozwoju RJG zalecana jest dieta ubogotłuszczowa z ograniczeniem mięsa czerwonego, z dodatkiem otrąb pszennych i bogata w błonnik (21).
Prewencja farmakologiczna
Istnieją doniesienia, że do leków hamujących karcinogenezę RJG należą: aspiryna, sulindac, piroxicam, wapń, witamina C. Przydatność tych preparatów w zapobieganiu HNPCC jest prawdopodobna, jednak jak dotąd nieudowodniona (20, 22, 23).
Kolonoskopia
Pełna kolonoskopia zalecana jest, co 1-2 lata poczynając od 20-25 r.ż. W rodzinach, w których RJG wystąpił w młodszym wieku kolonoskopie należy rozpocząć wykonywać o 5 lat wcześniej od wieku najmłodszej osoby z RJG. W przypadkach, w których w trakcie endoskopii nie można było dokładnie ocenić całego jelita wskazane jest wykonanie wlewu kontrastowego (20, 24).
Diagnostyka guzów pozaokrężniczych
W związku ze zwiększoną częstością występowania w rodzinach z HNPCC nowotworów narządu rodnego, u kobiet zalecane są od 35 r.ż. coroczne szczegółowe badania ginekologiczne z dopochwowym USG i histopatologicznym badaniem wyskrobin z jamy macicy włącznie. Ponadto w części przypadków wskazane są badania ukierunkowane na wykrywanie innych nowotworów częściej występujących w danej rodzinie (np. żołądek, układ moczowy, pierś) (5, 20).
Chirurgia
Polipektomia endoskopowa zalecana jest w przypadku polipów łagodnych, nienawrotowych. Natomiast u pacjentów z gruczolakami: mnogimi i/lub – nawrotowymi i/lub – o dużym stopniu dysplazji i/lub – kosmkowymi, należy rozważyć profilaktyczną kolektomię (24). Przeważa pogląd, że nie jest wskazana profilaktyczna chirurgia u pacjentów bez zmian patologicznych jelita grubego nawet wówczas, gdy osoby badane są nosicielami zmutowanych genów dla HNPCC (20). Stwierdzenie wysokiego odsetka guzów synchronicznych (u ponad 15% chorych w chwili pierwotnej diagnozy) oraz metachronicznych (ok. 45% pojawia się w ciągu 10 lat od usunięcia pierwotnego guza) zdecydowało, iż zarówno w profilaktycznej chirurgii jak i u chorych z rodzin z HNPCC z rozpoznanym histopatologicznie RJG polecane są następujące rodzaje zabiegów chirurgicznych (20, 24):
1. proktokolektomia z ileostomią;
2. kolektomia z zespoleniem ileo-rektalnym;
3. proktokolektomia z ileoanalnym „pouchem” – S, J, W lub H.
Pierwszy z proponowanych zabiegów jest wprawdzie najbardziej radykalny, pozbywamy się bowiem w całości śluzówki jelita grubego, a zatem i ryzyka nawrotu, ale zabieg ten oznacza równocześnie ciężkie okaleczenie często połączone z zaburzeniami w oddawaniu moczu i dysfunkcją seksualną.
Kolektomia z zespoleniem ileo-rektalnym chroni od tego rodzaju powikłań wymaga jednak częstej kontroli pozostawionego odcinka odbytnicy z uwagi na ryzyko nawrotu.
Proktokolektomia z ileoanalnym „pouchem” S, J, W lub H jest metodą mającą dopiero historię kilkunastoletnią stąd trudno o jej jednoznaczną ocenę.
U kobiet z zespołem Lyncha operowanych z powodu RJG i będących w wieku okołomenopauzalnym i starszym, ze względu na zwiększone ryzyko rozwoju raków trzonu macicy i jajnika zalecane jest poszerzenie zabiegu o wycięcie macicy wraz z przydatkami (20, 25).
Wszystkie te zabiegi są bardziej rozległe od zabiegów klasycznych stosowanych w leczeniu RJG i cechuje je zwiększona częstość ww. powikłań. Mimo to są one zalecane w leczeniu HNPCC, ponieważ nadrzędnym problemem u chorych z zespołem Lyncha jest wysokie ryzyko rozwoju drugiego pierwotnego ogniska RJG.
Wykazano już, że zastosowanie odpowiednich programów opieki medycznej w rodzinach z HNPCC zapewnia zwiększoną wykrywalność wczesnych, bezobjawowych RJG. Ponadto badania prospektywne potwierdziły założenie, iż dzięki odpowiedniemu postępowaniu zmniejsza się również zachorowalność na RJG – ryzyko spada z ok. 80 do 30% oraz większy jest odsetek wyleczeń i czas przeżycia chorych z RJG w rodzinach z zespołem Lyncha. Odpowiednio prowadzony nosiciel mutacji MSH2/MLH1 nie powinien umrzeć z powodu RJG (26). Piśmiennictwo
1. Lovett E: Family studies in cancer of the colon and rectum. Br J Surg 1976, 63: 13-18.
2. Lynch HT, Lynch J, Lynch P: Management and control of familial cancer. In: Mulvill JJ, Miller RW, Fraumeni JF, eds. Genetics of Human Cancer. New York, Raven Press, 1977, 3: 235-55.
3. Ponz de Leon M, et al.:. Familial aggregation of tumors in the three-year experience of a Population-based Colorectal Cancer Registry. Cancer Research 1989, 49: 4344-8.
4. Lynch HT, et al.: Hereditary colorectal cancer. Seminars in Oncology 1991, 18: 337-66.
5. Vasen H: Inherited forms of colorectal, breast, and ovarian cancer. Surgical Oncology Clin. N-Am. 1994, 3: 501.
6. Lynch HT, Krush AJ: Cancer family „G” revisited: 1895 – 1970. Cancer 1971, 27: 1505-11.
7. Fishel R, et al.: The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75: 1027-38.
8. Leach FS, et al.: Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993, 75: 1215-25.
9. Nicolaides NC, et al.: Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994, 371: 75-80.
10. Papadopoulos N, et al.: Mutation of a mutL homolog in hereditary colon cancer. Science 1994, 263: 1625-9.
11. Vasen HF, et al.: The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991, 34: 424-5.
12. Vasen HFA, et al.: New Clinical Criteria for Hereditary Nonpolyposis Colorectal Cancer (HNPCC, Lynch syndrome) Proposed by the International Collaborative Group on HNPCC. Gastroenterology 1999, 116: 1453-6.
13. Rodriques-Bigas MA, et al.: A National Cancer Institute Workshop on Hereditary on-polyposis Colorectal Cancer Syndrome: meeteng highlights and Bethesda Guidelines. J. Nat Cancer Ist 1997, 89: 1758-62.
14. Park JG, et al.: Suspected Hereditary Nonpolyposis Colorectal Cancer. Dis Colon Rectum 1999, 42: 710-6.
15. Park JG, et al.: Suspected HNPCC and Amsterdam criteria II: evaluation of mutation detection rate, an international collaborative study. Int. J. Colorectal Dis 2002, 17: 109-14.
16. Kladny J, et al.: Nuclear pedigree criteria of suspected HNPCC. Her Can in Clin Pract 2003, 1: 34-8.
17. Kurzawski G, et al.: Germline MSH2and MLH1mutational spectrum including large rearrangements in HNPCC families from Poland (update study). Clin Genet 2006, 69: 40-7.
18. Kurzawski G, et al.: Mutation analysis of MLH1and MSH2genes performed by denaturing high-performance liquid chromatography. J Biochem Biophys Meth 2002, 51: 89-100.
19. Vasen HF, et al.: Surveillance in Hereditary Nonpolyposis Colorectal Cancer: an international cooperative study of 165 families.The International Collaborative Group on HNPCC. Dis Colon Rectum 1993, 36: 1-4.
20. Lynch H, Lynch J: Lynch syndrome: Natural history, Genetic Counseling and Prevention. J Clin Oncol 2000, 18: 19-31.
21. Willet W, et al.: Relation of meat, fat, and fiber intake to the risk of colon cancer in a prospective study among women. N Engl J Med 1990, 323: 1664-72.
22. Bralow SP: Primary and secondary chemoprevention of colorectal cancer: Hereditary colorectal cancer. Springer Verlag Tokyo 1990, 231.
23. Muscat JE, Stellman SD,Wynder EL: Nonsteroidal antiiflamatory drugs and colorectal cancer. Cancer 1994, 74: 1847.
24. Vasen HF, Nagengast FM, Khan PM: Interval cancers in hereditary non-polyposis colorectal cancer (Lynch syndrome). Lancet 1995, 345: 1183-4.
25. Watson P, et al.: The risk of endometrial cancer in hereditary nonpolyposis colorectal cancer. Am J Med 1994, 96: 516-20.
26. Järvinen HJ, et al.: Controlled 15-year Trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 2000; 118: 829-34.
otrzymano/received: 2008-04-23 zaakceptowano/accepted: 2008-05-15 Adres/address: *Józef Kładny Klinika Chirurgii Ogólnej i Onkologicznej Pomorska Akademia Medyczna al. Powstańców Wielkopolskich 72, 70-111 Szczecin tel.: (0-91) 466-11-60 e-mail: jkladny@sci.pam.szczecin.pl Pełna wersja artykułu Zespół Lyncha (HNPCC) dostępna w Czytelni Medycznej Borgis. |
  Proszę kliknąć w wybraną okładkę aby przejść na stronę czasopisma
 |






Chcesz być na bieżąco? Polub nas na Facebooku: strona Wydawnictwa na Facebooku |