|
© Borgis - Postępy Nauk Medycznych 2, s. 64-68
*Wojciech Zgliczyński
Postępy w rozpoznawaniu i leczeniu akromegalii
Progress in diagnosis and treatment of acromegaly
Klinika Endokrynologii Centrum Medycznego Kształcenia Podyplomowego w Warszawie
Kierownik Kliniki: prof. CMKP dr hab. med. Wojciech Zgliczyński Streszczenie
W artykule przedstawiono aktualne zasady rozpoznawania i leczenia akromegalii, zwracając uwagę na postępy w tej dziedzinie, które związane są z: – wprowadzeniem kryteriów rozpoznawania choroby, polegających na wykazaniu zwiększonego stężenia GH> 2,5 ?g/l przy braku zahamowania wydzielania GH ≤ 1,0 ?g/l po doustnym podaniu 75 g glukozy oraz podwyższonego stężenia IGF-1 powyżej normy dla płci i wieku. – przedoperacyjnym zastosowaniem analogów somatostatyny o przedłużonym działaniu, które poprawiają wyniki leczenia neurochirurgicznego, szczególnie w przypadkach dobrze reagujących na leczenie farmakologiczne. – wprowadzeniem analogów somatostatyny o przedłużonym działaniu oraz antagonistów receptora GH, co przyczynia się do dobrej kontroli przebiegu choroby u pacjentów po nieskutecznej operacji gruczolaka somatotropowego przysadki i u osób, które takiemu zabiegowi poddać się nie mogą. Słowa kluczowe: akromegalia, GH, IGF-1, analogi somatostatyny
Summary
We present actual guidelines for acromegaly diagnosis and treatment, with special attention on recent developments in clinical practice: – Implementation of strict diagnosis criteria: increased random GH level in cases of lack GH suppression after oral glucose load and increased IGF-1 level, – Pre-operative medical treatment with long-action somatostatin analogues improving neurosurgery outcome, especially in good-responders for somatostatin receptor ligands. – Medical therapy with long-acting somatostatin analogues and growth hormone antagonist – improving disease control in cases of transsphenoideal neurosurgery failure or contraindications. Key words: acromegaly, GH, IGF-1, somatostatin analogs
Definicja
Akromegalia klinicznie charakteryzuje się powiększeniem twarzoczaszki, dłoni i stóp, rozrostem tkanek miękkich, kości oraz narządów wewnętrznych w następstwie nadmiernego wydzielania hormonu wzrostu (GH) przez gruczolak przysadki wywodzący się z komórek somatotropowych (1).
Rys historyczny
W 1886 roku francuski neurolog Pierre Marie nabyty rozrost twarzy, rąk i stóp nazwał akromegalią. Rok później Oskar Minkowski w Berlińskim Tygodniku Lekarskim zasugerował związek przyczynowy między powiększeniem przysadki a tą chorobą. A już w 1909 roku amerykański neurochirurg Harvey Cushing wykazał ustąpienie somatycznych objawów akromegalii po operacyjnym usunięciu guza przysadki.
Epidemiologia
Częstość występowania akromegalii określa się na 50 do 70 przypadków na milion. W skali rocznej rozpoznaje się 4 nowe zachorowania na milion (2). Zgodnie z tymi wyliczeniami w naszym kraju na akromegalię winno chorować około 2 tys. osób. Śmiertelność pacjentów z akromegalią jest znacznie większa, a czas przeżycia krótszy średnio o 10 lat w porównaniu z ogólną populacją. Najczęstszą przyczyną zgonu są choroby układu sercowo-naczyniowego, gdzie śmiertelność jest 2 do 3 razy większa niż w pozostałej populacji (3). Akromegalię stwierdza się dwukrotnie częściej u kobiet niż u mężczyzn.
Etiopatogeneza
Główną przyczyną nadmiernego wydzielania GH, prowadzącego do objawów akromegalii są gruczolaki przysadki wywodzące się z komórek somatotropowych. Są to guzy pochodzenia monoklonalnego, powstałe w wyniku mutacji pojedynczej komórki. Okazuje się, że przyczyną niekontrolowanego wzrostu komórek gruczolaka produkującego GH u 40% chorych jest jedna z dwóch mutacji punktowych podjednostki α białka Gs, doprowadzająca do ustalenia receptora dla GH-RH w stanie ciągłej aktywacji. Prowadzi to do wzmożonej proliferacji zmienionych genetycznie komórek somatotropowych i niekontrolowanego wydzielania hormonu wzrostu (4). Niezwykle rzadko (<1% przypadków) może być następstwem ektopowego wydzielania GH przez guzy neuroendokrynne (5), bądź zwiększonego wydzielania GH z przerośniętych komórek somatotropowych przysadki pobudzanych przez somatoliberynę (GHRH) ektopowo wydzielaną przez rakowiak (carcinoid) oskrzeli lub przewodu pokarmowego albo wyspiak trzustki (6). Nadmierne wydzielanie hormonu wzrostu prowadzi do zwiększonej syntezy w wątrobie i tkankach obwodowych somatomedyn, głównie somatomedyny C, inaczej insulinopodobnego czynnika wzrostowego-1 – IGF-1 ( Insulin-like Growth Factor-1). IGF-1 pobudza układy enzymatyczne komórek stymulując ich podziały kariokinetyczne warunkujące rozrost tkanek miękkich i kości. U dzieci i młodzieży z niezakończonym procesem wzrastania, u których nie doszło jeszcze do zrośnięcia nasad kości długich z trzonami, zwiększone stężenie IGF-1 prowadzi do ich nadmiernego wzrostu – gigantyzmu. U młodych pacjentów guzy wydzielające GH z reguły są większe i bardziej agresywne. W chwili rozpoznania 80% gruczolaków somatotropowych to makrogruczolaki, a ponad 70% z nich to guzy o średnicy przekraczającej 20 mm. W przewidywaniu charakteru i przebiegu klinicznego guza pomaga jego ocena za pomocą mikroskopii elektronowej. Gruczolaki o gęstym rozmieszczeniu ziarnistości wewnątrzwydzielniczych (rozpoznawane najczęściej po 50-tym roku życia) wykazują powolny wzrost, natomiast te z rzadko rozmieszczonymi ziarnistościami (rozpoznawane najczęściej u młodszych) z reguły przebiegają w sposób bardziej agresywny (7).
Objawy i przebieg choroby
Objawy akromegalii przedstawiono w tabeli 1.
Tabela 1. Objawy akromegalii.
Gruczolaki przysadki wywołujące akromegalię charakteryzują się umiarkowaną szybkością wzrastania a objawy związane z ekspansją guza i uciskiem na skrzyżowanie nerwów wzrokowych pojawiają się po kilku latach trwania choroby. Początkowo obserwuje się stopniowo postępujące zmiany wyglądu: powiększenie doni i stóp, twarzoczaszki i języka, obrzmienie tkanek miękkich oraz charakterystyczne zlewne poty i częste bóle głowy. Ze względu na długi okres czasu jaki z reguły upływa do rozpoznania akromegalii i podjęcia skutecznego leczenia chorzy narażeni są na długotrwałe oddziaływanie wysokich stężeń czynników wzrostowych – m.in. IGF-1, co jest przyczyną pojawienia się zmian, które po latach prowadzą do śmierci (3, 7). Należy tu wymienić:
1) powikłania z zakresu układu sercowo-naczyniowego, nadciśnienie tętnicze, kardiomiopatia przerostowa i związane z nią: zaburzenia rytmu i niewydolność serca oraz udary mózgu;
2) skłonność do rozrostów nowotworowych: złośliwych – najczęściej jelita grubego, ale też sutków, nerek, tarczycy i prostaty, oraz łagodnych – guzków tarczycy, polipów jelita grubego, mięśniaków macicy, łagodnego przerostu prostaty;
3) powikłania w układzie oddechowym w postaci zespołu bezdechu sennego i upośledzonej drożności górnych dróg oddechowych;
4) zaburzenia metaboliczne: nietolerancja glukozy związane z hiperinsulinizmem i jawną cukrzycą;
5) zmiany zwyrodnieniowo-wytwórcze w układzie kostno-stawowym (osteoartroza) – dające bóle i ograniczenie ruchomości w stawach;
6) zaburzenia z zakresu obwodowego układu nerwowego w postaci neuropatii, najczęściej występujący zespół cieśni nadgarstka,
Z podsumowanych ostatnio danych z 29 polskich ośrodków, dotyczących obserwacyjnego programu badawczego leczenia Sandostatin LAR 360 chorych z akromegalią wynika, że okres od pojawienia się pierwszych objawów do rozpoznania choroby wynosił średnio 8,8 lat. Na tym etapie choroby u 64,2% pacjentów pojawiło się nadciśnienie tętnicze, u 20,3% cukrzyca, a u 8,3% niewydolność serca. U 59,2% stwierdzono obecność wola, a u 8,6% nadczynność tarczycy (8).
Rozpoznanie
Rozpoznanie czynnej akromegalii opiera się na wykazaniu objawów klinicznych, hormonalnych i radiologicznych. Z objawów klinicznych o czynnym procesie akromegalii, poza charakterystycznymi objawami somatycznymi, świadczą utrzymujące się zlewne poty i bóle głowy. W diagnostyce hormonalnej podstawowe znaczenie ma wykazanie: podwyższonego stężenia GH w surowicy krwi oraz braku zahamowania wydzielania GH poniżej 1 μg/l (9) lub, według nowszych kryteriów, poniżej 0,4 ?g/l (10), po doustnym podaniu 75 g glukozy oraz podwyższonego stężenia IGF-1– powyżej normy dla płci i wieku (ryc. 1 – przedstawia algorytm postępowania). Z badań radiologicznych największe znaczenie ma wykazanie obecności gruczolaka przysadki w badaniu MR lub TK.
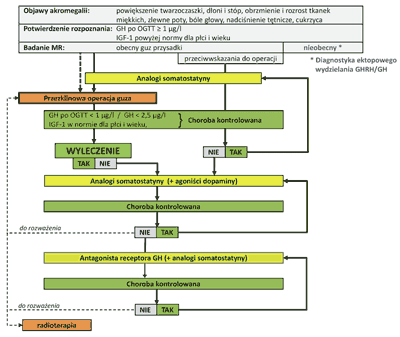 Ryc. 1. Algorytm postępowania w akromegalii.
W każdym przypadku akromegalii należy przeprowadzić badanie okulistyczne oceniając dno oczu (zaawansowanie zmian nadciśnieniowych) oraz pole widzenia. Zazwyczaj w momencie rozpoznania akromegalii wielkość guza odpowiada makrogruczolakowi (ponad 70%) z możliwością ekspansji ponadsiodłowej i uciskiem na skrzyżowanie nerwów wzrokowych (7).
Konieczne jest również wykluczenie niedoczynności przysadki (możliwość ucisku guza na zdrową część gruczołową przysadki) oraz pierwotnej nadczynności tarczycy (często występujące w akromegalii zmiany guzkowe tarczycy mogą ulegać autonomizacji). Pamiętać należy o możliwości hiperprolaktynemii (około 25% to guzy mieszane wydzielające również PRL lub uciskające na szypułę przysadki). Dodatkowo akromegalia może kojarzyć się z pierwotną nadczynnością przytarczyc i wyspiakami trzustki w zespole MEN 1.
Leczenie
Głównym celem leczenia akromegalii jest zmniejszenie związanej z podwyższonymi stężeniami GH i IGF-I nadmiernej śmiertelności w porównaniu do populacji ludzi zdrowych. Leczeniem z wyboru, prowadzącym do trwałego wyleczenia, jest operacyjne usunięcie guza przez zatokę klinową (ryc. 1). W przypadkach niewielkich gruczolaków, nie wykazujących ekspansji ponad przeponę siodła i do zatoki jamistej jest to stosunkowo łatwe dla wyspecjalizowanego w tego rodzaju zabiegach neurochirurga. Większy problem stanowią makrogruczolaki z zajęciem skrzyżowania nerwów wzrokowych, zatoki jamistej a czasami płatów skroniowych lub czołowych mózgu. Wydaje się, że optymalnym postępowaniem w tego rodzaju przypadkach jest przedoperacyjne zastosowanie analogów somatostatyny o przedłużonym działaniu (octreotydu – Sandostatin LAR firmy Novartis, lanreotydu – Somatuline PR lub ostatnio wprowadzonej formy Autogel firmy Ipsen), które u większości leczonych obniżają, a u około 50% normalizują stężenie GH. Istotnym jest, że leczenie prowadzi do zmniejszenia wielkości i zmiękczenia konsystencji guza. Im mniejszy guz i bardziej miękki tym łatwiejszy do całkowitego usunięcia przez zatokę klinową (ryc. 2).
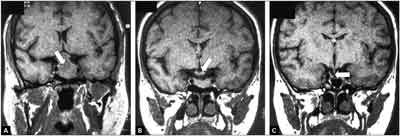 Ryc. 2. Obrazowanie okolicy soidła tureckiego w projekcji czołowej w badaniu MR u chorej z akromegalią.
Przykład istotnego zmniejszenia się makrogruczolaka przysadki po przedoperacyjnym zastosowaniu analogu somatostatyny. A. Obraz wyjściowy makrogruczolaka uciskającego skrzyżowanie wzrokowe (strzałka). B. Obraz po trzymiesięcznym leczeniu: zmniejszenie guza z uwolnieniem skrzyżowania wzrokowego (strzałka) – co zwiększa szansę doszczętności operacji. C. Obraz po doszczętnej operacji guza; uwidoczniona szypuła łączy się z małą częścią gruczołu przysadki (strzałka). Rokowanie co do wyleczenia na drodze operacyjnej zależy od wielkości guza przysadki oraz doświadczenia neurochirurga. Przyjmuje się, że skuteczność operacyjnego leczenia akromegalii w wyspecjalizowanych ośrodkach waha się od 80% w przypadku mikrogruczolaków do mniej niż 50% w przypadku guzów o średnicy>1 cm (7). Z doświadczenia autora wynika, że przedoperacyjne zastosowanie analogów somatostatyny istotnie zwiększa skuteczność leczenia neurochirurgicznego (11).
W przypadku braku zgody pacjenta na leczenie operacyjne lub wobec przeciwwskazań do tego typu leczenia stosuje się przewlekle analogi somatostatyny o przedłużonym działaniu. Podobną formę przewlekłego leczenia zachowawczego należy prowadzić w przypadkach nie wyleczonych operacyjnie lub nie rokujących wyleczenia skutecznym zabiegiem neurochirurgicznym. Celem terapii chorych z akromegalią analogami somatostatyny o przedłużonym działaniu jest utrzymanie stężenia GH w surowicy poniżej 2,5 ?g/l, a po doustnym obciążeniu glukozą <1 ?g/l i zmniejszenie stężenia IGF-I do wartości prawidłowych dla płci i wieku (9). Wymienione wartości GH i IGF-I świadczą o prawidłowej kontroli przebiegu choroby, co jest warunkiem zmniejszenia nadmiernej umieralności chorych z akromegalią (13, 14). W przypadku braku skuteczności analogów somatostatyny należy dołączyć do leczenia agonistę dopaminy, przeprowadzić reoperację guza, zastosować antagonistę receptora GH bądź w ostateczności rozważyć jego radioterapię (ryc. 1). Leczenie promieniami jonizującymi z zastosowaniem energii megavoltowej należy stosować we frakcjonowanych dawkach nie większych niż 190 cGy dziennie. Całkowita dawka nie powinna przekraczać 4500 cGy. Stosowanie większych dawek wiąże się z dużym prawdopodobieństwem popromiennych powikłań, takich jak: nieodwracalna niedoczynność przysadki, utrata słuchu, wzroku lub nawet życia (15). Pamiętać należy, że efekty radioterapii w postaci normalizacji stężenia GH pojawiają się po kilku latach od zakończenia terapii.
Za ograniczeniem tej formy leczenia przemawiają również dane wskazujące na wzrost śmiertelności chorych z akromegalią po przebytej radioterapii (16).
Od niedawna dostępna jest nowa opcja leczenia farmakologicznego antagonistą receptora GH – pegvisomantem. Pegvisomant jest analogiem GH, który różni się od naturalnego hormonu kilkoma resztami aminokwasowymi. Podstawienie określonych reszt aminokwasowych w miejscu wiązania z receptorem sprawia, że aktywacja receptora GH zostaje zablokowana, czego skutkiem jest zahamowanie syntezy i działania IGF-1 (17). Badania kliniczne wskazują, że do 97% pacjentów leczonych pegvisomantem osiąga normalizację osoczowego stężenia IGF-1 (19).
Podsumowanie
Postępy w rozpoznawaniu i leczeniu akromegalii związane są z:
– Wprowadzeniem kryteriów rozpoznawania choroby, polegających na wykazaniu zwiększonego stężenia GH> 2,5 ?g/l przy braku zahamowania wydzielania GH ≤ 1,0 ?g/l po doustnym podaniu 75 g glukozy oraz podwyższonego stężenia IGF-1 powyżej normy dla płci i wieku.
– Przedoperacyjnym zastosowaniem analogów somatostatyny o przedłużonym działaniu, które poprawiają wyniki leczenia neurochirurgicznego, szczególnie w przypadkach dobrze reagujących na leczenie farmakologiczne.
– Wprowadzeniem analogów somatostatyny o przedłużonym działaniu oraz antagonistów receptora GH, co przyczynia się do dobrej kontroli przebiegu choroby u pacjentów po nieskutecznej operacji gruczolaka somatotropowego przysadki i u osób, które takiemu zabiegowi poddać się nie mogą. Piśmiennictwo
1. Zgliczyński W: Guzy przysadki. [W]: Szczeklik A., red.: Choroby wewnętrzne. Wyd. 1 Medycyna Praktyczna, Kraków, 2005; 1021-32.
2. Paisley A, Trainer PJ: Recent developments in the therapy of acromegaly. Expert Opin. Investig. Drugs 2006; 15(3): 251-256.
3. Orme SM, et al.: Mortality and cancer incidence in acromegaly: a retrospective cohort study. United Kingdom Acromegaly Study Groups. J. Clin. Endocrinol. Metab., 1998; 83: 2730-34.
4. Landis CA, et al.: GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989; 340: 692-6.
5. Melmed S, et al.: Acromegaly due to secretion of growth hormone by an ectopic pancreatic islet-cell tumor. N. Engl. J. Med., 1985; 312: 9-17.
6. Thorner MO, et al.: Somatotroph hyperplasia: successful treatment of acromegaly by removal of a pancreatic islet tumor secreting a growth hormone-releasing factor. J. Clin. Invest., 1982; 70: 965-77.
7. Melmed S: Acromegaly. N. Engl. J. Med., 2006; 355: 2558-73.
8. Zgliczyński W: Obserwacyjny program badawczy leczenia Sandostatin LAR chorych z akromegalią (CSMS996BPL01) – podsumowanie - Endokrynol. Pol., 2007; 58(6) – w druku.
9. Giustina A, et al.: Criteria for cure of acromegaly: a consensus statement. J. Clin. Endocrinol. Metab., 2000; 85: 526-29.
10. Melmed S, et al.: Consensus steatement: medical management of acromegaly. Eur. J. Endocrinol., 2005; 153: 737-40.
11. Zgliczyński W, et al.: The somatostatin analog (SR-Lanreotide) preatreatment improves surgical outcome in acromegalic patients harbouring pituitary tumours with somatostatin receptors. J. Endocrinol. Invest., 1997; 20(7): 45-47.
12. Bolanowski M, et al.: Konsensus Polskiego Towarzystwa Endokrynologicznego. Przygotowanie analogami somatostatyny do leczenia operacyjnego akromegalii. Endokrynol. Pol., 2007; 58(4): 350-353.
13. Bates AS, et al.: An audit of outcome of treatment in acromegaly. Q J Med., 1993; 86: 293-99.
14. Rajasoorya C, et al.: Determinaty of clinical outcome and survival in acromegaly. Clin. Endocrinol., (OXF) 1994; 41: 35-102.
15. Zgliczyński W, et al.: Uszkodzenie mózgu po radioterapii guzów przysadki. Endokrynol. Pol., 2003; 54: 536-544.
16. Ayuk J, et al.: Growth hormone and pituitary radiotherapy, but not serum insulin-like growth factor-1 concentrations, predict excess mortality in patients with acromegaly. J. Clin. Endocrinol. Metab., 2004; 89: 1613-17.
17. Trainer PJ, et al.: Treatment of acromegaly with the growth hormone – receptor antagonist pegvisomant. N. Engl. J. Med., 2000; 342: 1171-7.
18. Zgliczyński W, Zdunowski P: Pegvisomant – antagonista receptora hormonu wzrostu w leczeniu akromegalii. Endokrynol. Pol., 2007; 58(5): 408-416.
19. Van der Lely J, et al.: Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet 2001; 358(9295): 1754-59.
otrzymano/received: 2007-11-28 zaakceptowano/accepted: 2007-12-29 Adres/address: *Wojciech Zgliczyński Klinika Endokrynologii CMKP, Szpital Bielański ul. Cegłowska 80, 01-809 Warszawa tel./fax: (0-22) 834-31-31 e-mail: klinendo@cmkp.edu.pl Pełna wersja artykułu Postępy w rozpoznawaniu i leczeniu akromegalii dostępna w Czytelni Medycznej Borgis. |
  Proszę kliknąć w wybraną okładkę aby przejść na stronę czasopisma
 |






Chcesz być na bieżąco? Polub nas na Facebooku: strona Wydawnictwa na Facebooku |