|
© Borgis - Postępy Nauk Medycznych 4, s. 282-288
*Paweł P. Liberski1, Beata Sikorska2, Caspar Jansen3, James W. Ironside4
Creutzfeldt-Jakob disease in old age
Choroba Creutzfeldta-Jakoba w późnym wieku
1,2Department of Molecular Pathology & Neuropathology, Medical University Lodz, Poland
Head: prof. Paweł P. Liberski 3Dutch Surveillance Centre for Prion Diseases, University Medical Centre Utrecht, Department of Pathology, Utrecht, The Netherlands 4National CJD Surveillance Unit, University of Edinburgh Western General Hospital, Edinburgh, UK Streszczenie
W pracy omówiono problemy choroby Creutzfeldta-Jakoba (CJD) w wieku podeszłym. Szybkie przeszukanie austriackiej bazy danych wykazało 17 przypadków pomiędzy 70.-75. rokiem życia; 10 przypadków: 76.-79. rok życia; 11 przypadków: 80.-85. rok życia, ale tylko jeden przypadek w 86. roku życia. W Holandii, ze 150 zarejestrowanych przypadków CJD, w latach 1998-2008, 39 występowało pomiędzy 70. a 75. rokiem życia (lub 28 jeśli 70. rok życia nie jest włączony do statystyki); 15 przypadków: 76.-79. rok życia; 9 przypadków: 80.-85. rok życia i żaden w 86. roku życia lub powyżej. W UK, spośród 1089 chorych na sporadyczną postać CJD, 42% odnotowano u osób powyżej 70. rokiem życia pomiędzy majem 1990 r. a listopadem 2009 r., obejmując 33% przypadków pomiędzy 70. a 79. rokiem życia i 9% przypadków powyżej 80. roku życia. Analiza polimorfizmu kodonu 129 genu kodującego białko prionu wykazała, że 63% przypadków sCJD to homozygoty Met Met; 18% homozygoty Met Val a 19% heterozygoty Met Val. W przypadkach powyżej 70. roku życia, rozkład ten zmienia się – 68 & Met Met, 16% Val Val i 16% Met Val. Dane polskie są szczególnie intersujace, 6 z 30 przypadków CJD wystapiła powyżej 70. roku życia (20%), stan kodonu 129 był dostępny dla trzech, 2 x Met Met i 1 x Met Val. Słowa kluczowe: choroba Creutzfeldta-Jakoba, priony, późny wiek
Summary
This review covers the problem of Creutzfeldt-Jakob disease (CJD) in the old age. A short inquiry performed for the purpose of this review revealed that in the Austrian database, there were 17 cases between 70-75 years old; 10 cases: 76-79 years old; 11 cases 80-85 years old but only 1 case 86 years old. In the Netherlands, out of a total of 150 CJD patients for years 1998-2008, they were 39 cases between 70 and 75 years of age (or 28 if age 70 is not included); 15 cases: 76-79 years of age; 9 cases: 80-85 years of age but no for 86 year of age or older. In the UK, 42% of 1089 sCJD patients were aged 70 or over at death during the period May 1990-November 2009, comprising 33% of sCJD cases aged 70-79 and 9% of cases aged 80 or over. Analysis of the codon 129 polymorphism in the prion protein gene ( PRNP) in the UK sCJD cases found overall 63% were methionine homozygotes (MM), 18% were valine homozygotes (VV) and 19% were heterozygotes (MV). In the cases age 70 or over at death, the distribution was 68% MM, 16% VV and 16% MV. The Polish data are very interesting in this respect: there are 6 of 30 cases of sCJD over 70 (20%); the 129 codon data are available for 3, 2 x MM, 1 x MV. The literature on the subject of this review is very limited. Key words: Creutzfeldt-Jakob disease, old age, prion diseases
Introduction
The transmissible spongiform encephalopathies (TSEs) or prion diseases are a group of neurodegenerative disorders which include kuru (1), Creutzfeldt-Jakob disease (CJD) (2), Gerstmann-Sträussler-Scheinker (GSS) disease (3), and fatal familial insomnia in man (4, 5) natural scrapie in sheep, goats (6-9), and mufflons (10), transmissible mink encephalopathy in ranch-reared mink (11), chronic wasting disease of deer, elk and moose in the USA and Canada (12-16), bovine spongiform encephalopathy or "mad cow disease” (17) and its analogues in several exotic species of antelopes (18-21), and wild felids in Zoological gardens (22), and feline spongiform encephalopathy in domestic cats (23).
These disorders are caused by a still not completely understood pathogen variously referred to as a "prion” or a slow, unconventional or atypical virus, or "virino” (24-27). Despite wide acceptance for the prion theory, these designations still reflect different views about the real molecular structure of the pathogen and, by the same token, our ignorance of its nature. Those who prefer to view this pathogen as composed "predominantly or exclusively” of a pathologically folded (misfolded) protein (PrPres, PrPSc; Sc, from scrapie or PrPd; d, from disease; PrPTSE, TSE from transmissible spongiform encephalopathy), use the term "prion” (28-31); hence the term "prion diseases” (25, 32, 33).
In 1995 Budka et al. (34) published a set of criteria to diagnose human TSEs on the basis of classical neuropathological staining and immunohistochemistry.
1. Creutzfeldt-Jakob disease
1.1. Spongiform encephalopathy in cerebral/or cerebellar cortex and/or subcortical gray matter
1.2. Encephalopathy with PrP immunoreactivity (plaque and/or diffuse synaptic and/or patchy/perivacuolar types)
2. Gerstmann-Sträussler-Scheinker disease (GSS). Encephalomyelopathy with multicentric PrP plaques
3. Fatal familial insomnia. Thalamic degeneration with focal cerebral spongiform change
4. Kuru (in the Fore population of New Guinea)
sCJD is a disease of the elderly, however, the number of deaths in persons over age 70 and higher is gradually rising. In the early 1970s, it was around 1 per year and in recent days, it is around 30 per year (35). Furthermore, the meadian ages of cases during time periods 1970-89, 1990-95 and 1996-2008 also increased from 64 years, through 66 years to 68 years, respectively. A short inquiry performed for the purpose of this review revealed that in the Austrian database, there were 17 cases between 70-75 years old; 10 cases: 76-79 years old; 11 cases 80-85 years old but only 1 case 86 years old. In the Netherlands, out of a total of 150 CJD patients for years 1998-2008, they were 39 cases between 70 and 75 years of age (or 28 if age 70 is not included); 15 cases: 76-79 years of age; 9 cases: 80-85 years of age but non for 86 year of age or older. In the UK, 42% of 1089 sCJD patients were aged 70 or over at death during the period May 1990-November 2009, comprising 33% of sCJD cases aged 70-79 and 9% of cases aged 80 or over (fig. 1). Analysis of the codon 129 polymorphism in the prion protein gene ( PRNP) in the UK sCJD cases found overall 63% were methionine homozygotes (MM), 18% were valine homozygotes (VV) and 19% were heterozygotes (MV). In the cases aged 70 or over at death, the distribution was 68% MM, 16% VV and 16% MV.
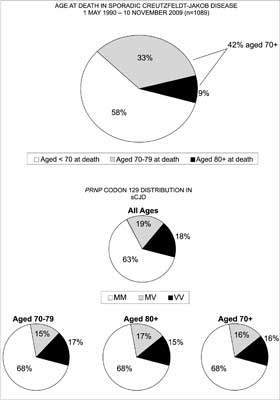 Fig. 1. (upper panel) Age at death in sCJD (1 May 1990 – 10 November 2009; n = 1089); (lower panel), PRNP codon 129 distribution.
The Polish data are very interesting in this respect: there are 6 of 30 cases of sCJD over 70 (20%); the 129 codon data are available for 3, 2 x MM, 1 x MV.
The literature on the subject of this review is very limited.
De Silva and colleagues (36) reviewed 12 cases over 80 years of age and added a case of 86-year-old CJD patient. The duration of disease for those 12 cases ranged between 1 and 5 months, the EEG showed PSWCs in 6 cases but was atypical in another six, and they presented as rather typical sCJD. The 86-year-old age case presented with disorientation and self-neglect, jerky limb movement and on EEG, slow background rhythms and periodic complexes. Terminally, she had frontal release signs. Neuropathological examination revealed spongiform change and immunoexpression of PrPSc.
The oldest case was published by Buganza et al. (37). This was a 98-year-old patient admitted to the hospital because of an ataxic gait of 10 days duration. The following day – myoclonic jerks were observed, followed by – generalized seizures. The EEG revealed continued seizure activity turned into periodic sharp wave complexes (PSWCs), otherwise typical for CJD. Western blot analysis revealed type 1 PrPSc, and molecular analysis revealed that the patient was a Methionine homozygote at the 129 codon of the PRNP gene. Neuropathological examination showed widespread spongiform change and diffuse PrP-immunoexpression.
There are several points to be raised by those very limited observations. First, all those cases presented as typical sCJD. The only possible exception is generalized seizures in the case of Buganza et al. (37). Of note, Collins et al. (38) described a case of 82-year-old woman who also presented with generalized seizures. However, that case was not only a methionine homozygote, but also a carrier of T188A mutation. Thus, it is tempting to speculate that seizures may be a sign of CJD in the elderly.
Those old CJD cases are also interesting from a pathogenetic perspective. According to the prion theory, sCJD may be caused by a stochastic event of a change of a normal PrP isoform into its pathologic counterpart. However, if this is really a stochastic event, it should be time-dependent, thus the number of sCJD cases should increase with age and definitively the reverse is observed (fig. 2-3), the number of sCJD cases in – old age is small.
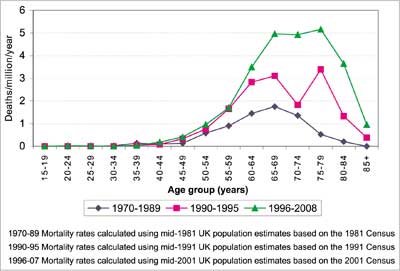 Fig. 2. Age-specific mortality rates from sporadic CJD in the UK 1970-2008. Courtesy of Prof. Robert Will, NCJDSU, Edinburgh, UK.
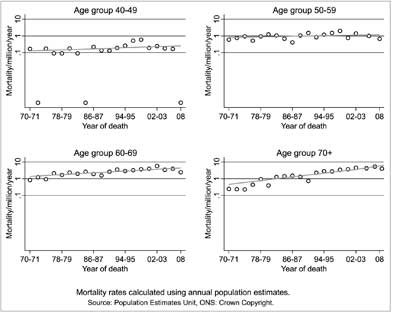 Fig. 3. Trends in mortality from sporadic CJD by age: 1970-2008. Courtesy of Prof. Robert Will, NCJDSU, Edinburgh, UK.
Variant CJD (vCJD) is worthy of separate consideration. vCJD is caused by a passage of BSE from bovine to humans and occurs in younger patients. However, Lorains et al. (39) described a unique vCJD case in a 74-year-old patient. He presented to the hospital with psychiatric symptoms, visual hallucinations and delusions of 1 month duration. His cognitive state declined, and he died with bilateral bronchopneumonia. At autopsy, abundant florid plaques, typical for vCJD were found in the brain and widespread PrPSc immunoreactivity was detected. As – for all definite vCJD cases this case was a methionine homozygote at codon 129 in the PRNP gene.
It is worthy to analyse those cases against a current sCJD classification based on the codon 129 status and the species of PrPSc (type 1 or 2 based on N-terminal cleavage site). Based on analysis of some 300 cases of sCJD, Parchi and colleagues (40), described 6 subtypes of sCJD that differ in some fundamental characteristics:
? MM1/MV1 or myoclonic/Heidenhain variants; age of onset 65.5 (42-91) for MV1 and age of onset 62.1 (51-72), for MV1
? VV2 – ataxic (or Betty Bronwell) variant; age of onset 61.3 (41-80)
? MV2 – kuru plaque variant, age of onset 59.4 (40-81)
? MM2-thalamic – thalamic variant or FFI, age of onset 52.3 (36-71)
? MM2-cortical, age of onset 64.3 (49-77)
? VV1, age of onset 39.3 (24-49)
This data clearly showed, that sCJD at the age over 70 may be seen in practically every subtype except perhaps the VV1 sCJD subtype; the classical MM1 and MV2 sCJD subtypes included cases of the highest age of onset (91 and 81, respectively), followed by MV1 classical sCJD variant (80 year at the onset). In the UK, the MM1 sCJD subtype was the commonest in all ages, including those aged 70 or over at death (fig. 4). This elderly group included all the recognised sCJD subtypes, including the rare VV1 subtype, cases with mixed PrP isoforms in all PRNP codon 129 genotypes and a case of the recently reported protease sensitive prionopathy (PSPr) (41, 42).
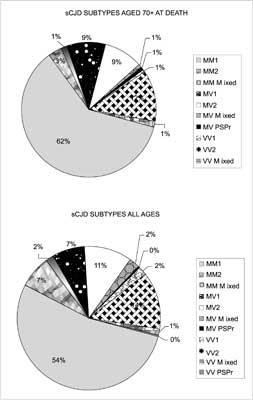 Fig. 4. sCJD subtypes in all ages and in the elderly.
As mentioned previously, seizures were stressed as signs perhaps overrepresented in elderly sCJD patients. They occur in 19% of the MM1 sCJD variant; 22% of MV2; 12% of MV1; none of VV1; 33% of MM2-cortical; none of MM2-thalamic; 11% of MV2 and 2% of VV2.
If we ranked sCJD subtypes according to the highest age of onset and compare seizures activity, we clearly see that there is no exact match between the highest age of onset and the fraction of cases with seizure. However, the youngest age groups (MM2-thalamic and VV1, the oldest age of onset – 71 and 49-year, respectively), demonstrated no seizures at all.
? MM1, 91 years of age; 19% of cases demonstrated seizures
? MV2, 81 years of age; 22% of cases demonstrated seizures
? VV2, 80 years of age; 2% of cases demonstrated seizures
? MV1, 72 years of age; 12% of cases demonstrated seizures
? MM2-cortical, 77 years of age; 33% of cases demonstrated seizures
? MM2-thalamic, 71 years of age; none of cases demonstrated seizures
? VV1, 49 years of age; none of cases demonstrated seizures
The major differential diagnosis of CJD in the old age group is Alzheimer disease (AD) and dementia with Lewy bodies (DLB). We (43-45) (fig. 5-6) and the others (46-49) showed Alzheimer Aβ-composed amyloid plaques in otherwise typical cases of sCJD and recently it has been demonstrated that PrP is one of the regulators of presenilins, proteins not only involved in a processing of βAPP into Aβ but also, when mutated, a cause of a familial AD linked to chromosome 14. However, in a systematic study of 110 neuropathologically verified CJD cases, Hainfellner et al. (50) found Alzheimer-type definite and probably CERAD AD changes in slightly over 10%, but in 19% of control brains. The authors concluded that Alzheimer-type neuropathology is merely age-related phenomenon.
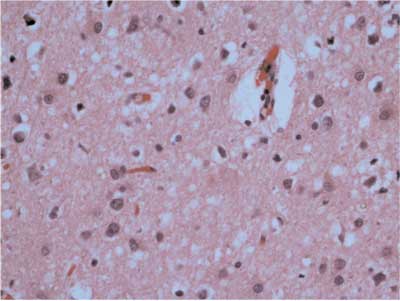 Fig. 5. A case of Creutzfeldta-Jakob disease; H & E staining, spongiform change is well visible. Amyloid plaque (not well visible), is marked with arrow.
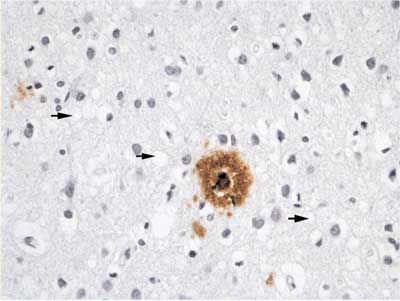 Fig. 6. The same case immunostained against Ab, amyloid plaque is visible in the center and spongiform change at the background (arrows).
However, in 2 cases of fCJD with E200K – 129M mutation (51), numerous Aβ immunoreactive plaques codistributed with spongiform change. Both cases were carriers of APOE ε4 allele and in non-carriers Aβ changes were not seen. Analogous Aβ-immunoreactive plaques were seen in a case of fCJD with V180I mutation (52). The most interesting in this respect is 28-year-old case of iCJD following dura matter grafting with abundant Aβ-immunoreactive plaques and amyloid angiopathy (53). Those changes met the CERAD criteria for definite AD. Because of the very young age, the concomitant AD and CJD is intriguing and suggest more that just a fortuitous change.
There are two sets of data suggesting a possible link between CJD and AD. First, the cellular isoform of PrP (PrPc) seems to regulate β-secretase cleavage of the βAPP (54). PrPc inhibits β-secretase and reduces Aβ formation. In contrast, depletion of PrPc by siRNA increases amount of Aβ. In mice with no PrP (129OlaPrP-/-), the amount of Aβ is also increased. In the context of AD changes observed in patients carrying PRNP gene mutation, two mutant forms of PrP: PG14 and A116V do not inhibit the β-secretase activity in vitro that leads to increase of the level of Aβ.
The second set of data concerns the putative role of the codon 129 homozygosity in the pathogenesis of AD. We (55) and the others (56) found the linkage between 129 codon homozygosity and the risk for AD. In conclusion, both AD and CJD may be interwoven.
The second disease to be discussed in a context of CJD in the old age is dementia with Lewy bodies (DLB) (57-61). Tschampa et al. (62) analysed a series of patients clinically suspected of CJD but in whom DLB was confirmed on the basis of neuropathology. In those 104 cases in whom CJD was ruled out, DLB was diagnosed in 14 cases. The sCJD cases were younger than DLB patients (median 64 years vs 72 years, respectively). DLB patients presented symptoms exclusively of dementia, dementia in CJD was accompanied by neurological signs or symptoms; myoclonic jerks, regarded as almost pathognomic for CJD, however, was present in 70% of DLB cases. In contrast, cerebellar signs, typically present in CJD, were not observed in DLB. To the contrary, fluctuations of the level of consciousness regarded as typical for DLB were found in 58% of DLB cases but in none of CJD cases. Visual hallucinations were also observed in 58% of DLB but also in 36% of CJD. The typical EEG with PSWCs were seen in 56% of CJD but only in 8% of DLB. The detection of 14-3-3 protein in the CSF was also discriminatory – it was observed in 56% cases of CJD but none of DLB patients. In conclusion, dementia, myoclonus and limb rigidity were common features of DLB and CJD while extrapyramidal features were the most common signs of DLB.
However, according to the Parchi et al. (40) classification, the MV2 subtype of sCJD demonstrates no typical EEG changes and the 14-3-3 protein is not present in the CSF. Thus, this variant may be more readily mistaken clinically for DLB.
In conclusion, CJD is a relatively rare disease in the elderly that resembles CJD in younger age groups. AD and DLB are likely clinical differentials, and autopsy is required to confirm the diagnosis and to allow subclassification of sCJD.
Professor Herbert Budka and Dr. Gabor Kovacs, Clinical Institute of Neurology, Medical University, Vienna, Austria, are kindly acknowledged for providing Austrian CJD data. Piśmiennictwo
1. Gajdusek DC, Gibbs CJ, Alpers MP: Experimental transmission of a kuru-like syndrome to chimpanzees. Nature 1966; 209: 794-796.
2. Gibbs CJ Jr, Gajdusek DC, Asher DM et al.: Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to chimpanzee. Science 1986; 161: 388-389.
3. Masters CL, Gajdusek DC, Gibbs CJ Jr: Creutzfeldt-Jacob disease virus isolations from the Gerstmann-Straussler syndrome. With an analysis of the various forms of amyloid plaque deposition in the virus inducted spongiform encephalopathies. Brain 1981; 104: 559-588.
4. Medori R, Montagna P, Tritschler HJ et al.: Fatal familial insomnia: a second kindred with mutation of prion protein gene at codon 178. Neurology 1992; 42: 669-671.
5. Medori R, Montagna P, Tritschler HJ et al.: Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N Engl J Med 1992; 326: 444-449.
6. M'Gowan JP: Investigation into the disease of sheep called "scrapie” (Traberkrankheit, La tremblante). With especial reference to its association with Sarcosporidiosis. William Blackwood and Sons, Edinburgh 1914.
7. Cuille J, Chelle PL: La "tremblante” du mouton est bien inoculable. C R Acad Sci (Paris) 1938; 206: 78-79.
8. Cuille J, Chelle PL: La tremblante du mouton est-elle determine par un virus filterable? C R Acad Sci (Paris) 1938; 11687-1688.
9. Culiie J, Chelle PL: La tremblante du mouton. Son etiologie. Rev Pathol Comp d'Hyg Gen 1938; 38: 1358-1372.
10. Wood JL, Lund LJ, Done SH: The natural occurrence of scrapie in moufflon. Vet Rec 1991; 130: 25-27.
11. Burger D, Hartsough GR: A scrapie-like disease of mink. [In:] Report of a Scrapie Seminar. United States Department of Agriculture 1964; paper 27: pp 225-227
12. Haigh JC, Mackintosh C, Griffin F: Viral, parasitic and prion diseases of farmed deer and bison. Rev Sci Tech 2002; 21: 219-248.
13. Liberski PP, Guiroy DC, Williams ES et al.: Deposition patterns of disease associated prion protein in captive mule deer brains with chronic wasting disease. Acta Neuropathol (Berl) 2001; 102: 496-500.
14. Williams ES, Miller MW: Chronic wasting disease in deer and elk in North America. Rev Sci Tech 2002; 21: 305-316.
15. Williams ES Young S: Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildlife Dis 1980; 16: 89-98.
16. Williams ES, Young S: Spongiform encephalkopathy in a Rocky Mountain elk. J Wildlife Dis 1982; 18: 465-471.
17. Wells GA, Scott AC, Johnson CT et al.: A novel progressive spongiform encephalopathy in cattle. Vet Rec 1987; 121: 419-420.
18. Cunnigham AA, Wells GAH, Scott AC et al.: Transmissible spongiform encephalopathy in greater kudu (Tragelaphus strepsiceros). Vet Rec 1993; 132: 68.
19. Fletwood AJ, Furley CW: Spongiform encephalopathy in an eland. Vet Rec 1990; 126: 215-222.
20. Jeffrey M, WellsGAH: Spongiform encephalopathy in a Nyala (Tragelaphus angasi). Vet Pathol 1988; 25: 398-399.
21. Kirkwood JK, Wells GA, Cunningham AA et al.: Scrapie-like encephalopathy in a greater kudu (Tragelaphus strepsiceros) which had not beed fed ruminant-derived protein. Vet Res 1992; 130: 365-367.
22. Willoughby K, Kelly DF, Lyon DG et al.: Spongiform encephalopathy in a captive puma (Felis concolor). Vet Res 1992; 131: 431-434.
23. Wyatt JM, Pearson GR, Smerdan T et al.: Spongiform encephalopathy in a cat. Vet Rec 1981; 129: 233-236.
24. Aguzzi A: Cell biology. Prion toxicity: all sail and no anchor. Science 2005; 308: 1420-1421.
25. Caughey B, Baron GS: Prions and their partners in crime. Nature 2006; 443: 803-810.
26. Liberski PP, Brown P: Prion diseases: from transmission experiments to structural biology – still searching for the cause. Folia Nauropathol 2004; 42 (Suppl A): 15-32.
27. Liberski PP Brown P: Disease-specific particles without prion protein diseases – phenomenon or epiphenomenon. Neuropathol Appl Neurobiol 2007; 33: 395: 397.
28. Prusiner SB: Some speculations about prions, amyloid, and Alzheimer's disease. N Engl J Med 1984; 310: 661-663.
29. Prusiner SB: Molecular biology of prion diseases. Science 1991; 252: 15151-1522.
30. Prusiner SB, Groth D, Serban A et al.: Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates of anti-PrP antibodies. Proc Natl Acad Sci USA 1993; 90: 10608-10612.
31. Prusiner SB, McKinley MP, Bowman KA et al.: Scrapie prions aggregate to form amyliod-like birefringent rods. Cell 1983; 35: 349-358.
32. Flechsig E, Weissmann C: The role of PrP in health and disease. Curr Mol Med 2004; 4: 337-353.
33. Weissmann C: The state of the prion. Nat Rev Microbio 2004; 2: 861-871.
34. Budka, Aguzzi A, Brown P et al.: Neuropathological diagnostic criteria for Creutzfeldt-Jacob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol 1995; 5(4): 459-66.
35. Creutzfeldt-Jacob disease surveillance in the UK. Seventeenth Annual Report 2008 of The National CJD Surveillance Unit, Western General Hospital, Edinburgh.
36. De Silva R, Findlay C, Awad J et al.: Creutzfeldt-Jacob disease in the elderly. Postgrad Med J 1997; 73: 557-559.
37. Buganza M, Ferrari S, Cecchini ME et al.: The oldest old Creutzfeldt-Jacob disease case. J Neurol Neurosurg Psychiatry 2009; 80: 1140-2.
38. Collins S, Boyd A, Fletcher A et al.: Novel prion protein gene mutation in an octogenarian with Creutzfeldt-Jacob disease. Arch Neurol 2000; 57 (7): 1058-63.
39. Lorains JW, Henry C, Agbamu DA et al. Variant Creutzfeldt-Jacob disease in an elderly patient. Lancet 2001; 357: 1339-40.
40. Parchi P, Giese A, Capellari S et al.: Classification of sporadic Creutzfeldt-Jacob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999; 46 (2): 224-33.
41. Gambetti P, Dong Z, Yuan J et al.: A novel human prion disease with abnormal prion protein sensitive to protease. Annals of Neurology 2008; 63: 697-708.
42. Head MW, Kinght R, Zeidler M et al.: A case of protease sensitive prionopathy in a patient in the United Kingdom. Neuropathology and Applied Neurobiology 2009 Aug 7 Epub ahead of print.
43. Barcikowska M, Mirecka B, Papierz W et al.: [A case of Alzheimer'disease simulating Creutzfeldt-Jacob disease] [Article in Polish]. Neurol Neurochir Pol 1992; 26(5): 703-10.
44. Barcikowska M, Kwieciński H, Liberski PP et al.: Creutzfeldt-Jacob disease with Alzheimer-type A beta-reactive amyloid plaques. Histopathology 1995; 26 (5): 445-50.
45. Liberski PP: Transmissible cerebral amyloidoses as a model for Alzheimer's disease. An ultrastructural perspective. Mol Neurobiol. 1994; 8 (1): 67-77.
46. Brown P, Jannotta F, Gibbs J et al.: Coexistence of Creutzfeldt-Jacob disease and Alzheimer's disease in the same patient. Neurology 1990; 40 (2): 226-8.
47. Powers JM, Liu Y, Hair LS et al.: Concomitant Creutzfeldt-Jacob and Alzheimer diseases. Acta Neuropathol 1991; 83 (1): 95-8.
48. Muramoto T, Kitamoto T, Koga H et al.: The coexistence of Alzheimer's disease and Creutzfeldt-Jacob disease in a patient with dementia of long duration. Acta Neuropathol 1992; 84(6): 686-9.
49. Tsuchiya K, Yagishita S, Ikeda K et al.: Coexistence of CJD and Alzheimer's disease: an autopsy case showing typical clinical features of CJD. Neuropathology 2004; 24 (1): 46-55.
50. Hainfellner JA, Wanschitz J, Jellinger K et al.: Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jacob disease. Acta Neuropathol 1998; 96 (2): 116-22.
51. Ghoshal N, Cali J, Pernin RJ et al.: Codistribution of amyloid beta plaques and spongiform degeneration in familial Creutzfeldt-Jacob disease with the E200K-129M haplotype. Arch Neurol 2009; 66 (10): 1240-6.
52. Yoshida H et al.: An autopsy case of Creutzfeldt-Jacob disease with a V108I mutation of the PrP gene and Alzheimer-type pathology. Neuropathology 2009 Aug23. [Epub ahead of print]
53. Preusser M, Ströbel T, Gelpi E et al.: Alzheimer-type neuropathology in a 28 year old patient with iatrogenic Creutzfeldt-Jacob disease after dural grafting. J Neurol Neurosurg Psychiatry 2006; 77 (3): 413-6.
54. Parkin ET, Watt NT, Hussain I et al.: Cellular prion protein regulates β-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc Natl Acad Sci USA, 2007; 26, 104 (26): 11062-7.
55. Golanska E, Hulas-Bigoszewska K, Rutkiewicz E et al.: Polymorphisms within the prion (PrP) and prion-like protein (Doppel) genes in AD. Neurology 2004; 62 (2): 313-5.
56. Del Bo R, Comi GP, Bresolin N et al.: Is M129V of PRNP gene associated with Alzheimer's disease? A case-control study and a meta-analysis. Neurobiol Aging 2006; 27 (5): 770.e1-770.e5. Epub 2005 Aug 15.
57. Haraguchi T, Seishi J, Hideki I et al.: Coexistence of Creutzfeldt-Jacob disease, Lewy body disease, and Alzheimer's disease pathology: an autopsy case showing typical clinical features of Creutzfeldt-Jacob disease. Neuropathology 2009; 29 (4): 454-9.
58. Koide T, Othake H, Nakajima T et al.: A patient with dementia with Lewy bodies and codon 232 mutation of PRNP. Neurology 2002; 59 (10): 1619-21.
59. Doran M, Larner AJ: EEG findings in dementia with Lewy bodies causing diagnostic confusion with sporadic Creutzfeldt-Jacob disease. Eur J Neurol 2004; 11 (12): 838-41.
60. Kraemer C, Lang K, Weckesser M et al.: Creutzfeldt-Jakob disease misdiagnosed as dementia with Lewy bodies. J Neurol 2005; 252 (7): 861-2. Epub 2005 Mar 17.
61. Vital A, Canron MH, Gil R et al.: A sporadic case of Creutzfeldt-Jacob disease with beta-amyloid deposits and alpha-synuclein inclusions. Neuropathology 2007; 27 (3): 273-7.
62. Tschampa HJ, Neumann M, Zern J et al.: Patients with Alzheimer's disease and dementia with Lewy bodies mistaken for Creutzfeldt-Jacob disease. J Neurol Neurosurg Psychiatry 2001; 71 (1): 33-9.
otrzymano/received: 2010-01-20 zaakceptowano/accepted: 2010-03-03 Adres/address: *Paweł P. Liberski Department of Molecular Pathology & Neuropathology, Medical University Łódź ul. Czechosłowacka 8/10, 92-216 Łódź e-mail: ppliber@csk.am.lodz.pl. Artykuł Creutzfeldt-Jakob disease in old age w Czytelni Medycznej Borgis. |
  Proszę kliknąć w wybraną okładkę aby przejść na stronę czasopisma
 |






Chcesz być na bieżąco? Polub nas na Facebooku: strona Wydawnictwa na Facebooku |