|
© Borgis - Postępy Nauk Medycznych 1, s. 69-74
*Beata Kasztelan-Szczerbińska1, Maria Słomka1, Jadwiga Daniluk1, 2, Krzysztof Celiński1, Halina Cichoż-Lach1, Mariusz Szczerbiński1, Agnieszka Zwolak2, Iwona Jastrzębska2
Komórki gwiaździste jako główny regulator sygnalizacji międzykomórkowej w procesie włóknienia wątroby
Stellate cells as a central regulator of intracellular signaling in the process of liver fibrosis
1Katedra i Klinika Gastroenterologii z Pracownią Endoskopową, Uniwersytet Medyczny w Lublinie
Kierownik Katedry i Kliniki: prof. dr hab. Maria Słomka 2Katedra Interny i Zakład Pielęgniarstwa Internistycznego, Uniwersytet Medyczny w Lublinie Kierownik Katedry: prof. dr hab. Jadwiga Daniluk Streszczenie
Włóknienie wątroby to rodzaj reakcji komórkowej na przewlekłe działanie czynnika uszkadzającego. Może ono prowadzić do marskości i niewydolności narządu z wszelkimi ich negatywnymi konsekwencjami – nadciśnieniem wrotnym z krwawieniami z przewodu pokarmowego, wodobrzuszem, zaburzeniami krzepnięcia i encefalopatią wątrobową. Komórki gwiaździste wątroby (hepatic stellate cells – HSC) to nie miąższowe komórki w fazie spoczynku, których główną funkcją w zdrowej wątrobie jest magazynowanie witaminy A. Pod wpływem uszkodzenia wątroby przechodzą one w fazę aktywacji, która polega na przemianie do fenotypu charakteryzującego się zdolnością do proliferacji, indukowania włóknienia i zapalenia oraz kurczliwością. Interakcje pomiędzy hepatocytami, komórkami Kupffera, komórkami zapalnymi i HSC wyzwalają sygnały profibrotyczne i prowadzą do włóknienia. Aktywacja HSC jest głównym zjawiskiem w procesie wątrobowej fibrogenezy, można ją podzielić na dwie główne fazy: 1. inicjację – zwaną również fazą przedzapalną i 2. fazę rozwinięcia, w której utrzymywany jest aktywny fenotyp HSC i w której postępuje włóknienie. Po usunięciu czynnika uszkadzającego dochodzi do redukcji liczby aktywnych HSC i rezolucji włóknienia. Do zjawiska tego dochodzi prawdopodobnie dwoma drogami: 1. przez powrót do fenotypu spoczynkowego lub 2. na drodze apoptozy. Wyniki badań przeprowadzone na przestrzeni ostatnich 20 lat wskazują na udział HSC nie tylko w procesach uszkodzenia wątroby, ale również w jej rozwoju, regeneracji, reakcjach na ksenobiotyk i immunoregulacji. Ogromny postęp wiedzy w tej dziedzinie naświetlił potencjalne możliwości dla interwencji terapeutycznej u pacjentów z przewlekłą chorobą wątroby. Nasza praca uwypukla charakterystykę komórek gwiaździstych oraz ich interakcje z innymi komórkami w okresie indukcji, progresji i rezolucji włóknienia w wątrobie. Słowa kluczowe: komórka gwiaździsta wątroby, włóknienie wątroby, macierz zewnątrzkomórkowa
Summary
Hepatic fibrosis represents cellular response to chronic liver injury. It can lead to cirrhosis and organ failure with all their negative consequences – portal hypertension with gastrointestinal bleeding, ascites, coagulopathy, and hepatic encephalopathy. Hepatic stellate cells (HSC) are nonparenchymal, quiescent cells, whose main function is to store vitamin A in normal liver. Upon liver injury they undergo an activation process, which represents a transition into proliferative, fibrogenic, proinflammatory and contractile phenotype. Cross – talk between hepatocytes, Kupffer cells, inflammatory cells and HSC induces profibrogenic signals and triggers fibrogenesis. The ativation of HSC remains a central event in the process of liver fibrosis and can be subdivided into two major phases: 1. initiation, also called pre-inflammatory stage and 2. perpetuation which maintain the activated phenotype of HSC and generate fibrosis. As the injurious agent is removed, the resolution of hepatic fibrosis with the decrease in activated HSC number can occur. Two potential pathways account for this process: 1. reversion to quiescent phenotype or 2. clearance through apoptosis. Research over past 20 years has indicated of HSC involvment not only in liver injury, but also in hepatic development, regeneration, xenobiotic responses and immunoregulation. Progress in this area has elucidated potential possibility of therapeutic intervention that might help patients with chronic liver disease. This review highlights characteristics of biology of hepatic stellate cells and their interaction with other cell types during induction, progession and resolution of liver fibrosis. Key words: hepatic stellate cell, liver fibrosis, extracellular matrix
Wprowadzenie
Badania przeprowadzone w ciągu ostatnich 20 lat potwierdziły, że komórki gwiaździste stanowią istotny element funkcjonowania wątroby oraz klucz do odpowiedzi tego narządu na bodźce uszkadzające. Powszechną uwagę badaczy skupiła aktywacja i przemiana tych komórek w miofibroblasty w procesie wątrobowej fibrogenezy. Postęp w poznaniu kolejnych ogniw tego fascynującego zjawiska był możliwy dzięki zastosowaniu nowoczesnych metod izolacji komórek, stworzeniu złożonych modeli genetycznych oraz udoskonaleniu narzędzi do badań i analiz naukowych, w tym cytometrii przepływowej, ilościowej metody polimerazowej reakcji łańcuchowej (real-time PCR), obrazowania konfokalnego oraz zastosowaniu molekularnych markerów do oznaczeń fenotypu i pochodzenia komórek.
Komórki gwiaździste zostały po raz pierwszy opisane przez Kupffera w 1876 roku. Obecność „sternzellen”(niemiecki termin dla komórki o gwiaździstym kształcie) potwierdził Rother w 1882 roku. Minęło dalszych 120 lat pośród różnorodności terminologii i jednoczasowego funkcjonowania w literaturze światowej wielu synonimów, takich jak komórki perisinusoidalne, komórki parasinusoidalne, wątrobowe perycyty, lipocyty, komórki magazynujące tłuszcze, komórki Ito. Dopiero w 1996 roku badacze uzgodnili jednolite mianownictwo i termin „komórka gwiaździsta” (hepatic stellate cell – HSC) stał się powszechnie obowiązującym (1, 2).
Komórki gwiaździste stanowią 1/3 część nieparenchymalnej populacji komórek i około 15% całkowitej liczby komórek rezydujących w zdrowej wątrobie (1). Zlokalizowane są w okołozatokowej przestrzeni Disse'go pomiędzy endotelium zatok i warstwą hepatocytów. Pozostają one w bezpośrednim kontakcie z zakończeniami nerwowymi, co potwierdza ich rolę w interakcjach neuro-humoralnych.
Cechą charakterystyczną komórek gwiaździstych w zdrowej wątrobie (HSC w stanie spoczynku) jest obecność w ich cytoplazmie kropelek zawierających witaminę A. Są one głównym jej magazynem. Do markerów, na podstawie których można rozpoznać HSC, należą: autofluorescencja witaminy A, okołozatokowa ich lokalizacja oraz ekspresja białek cytoskeletonu – desminy i kwaśnego, fibrylarnego białka glejowego (ekspresja obu białek może być jednak zmienna) (3, 4). W odpowiedzi na czynnik uszkadzający HSC tracą kropelki witaminy A, a następnie ulegają istotnej morfologicznej i funkcjonalnej przemianie zwanej procesem aktywacji. W jej trakcie nabywają cech fenotypowych zbliżonych do miofibroblastów oraz rozpoczynają intensywną syntezę kolagenu, głównie typu I (4, 5). HSC są kluczowym ogniwem w patogenezie włóknienia wątroby – indukują odkładanie i nieprawidłowy remodeling macierzy zewnątrzkomórkowej (6, 7, 8). Postępujące włóknienie prowadzi w konsekwencji do rozwoju poważnych powikłań schyłkowej choroby wątroby, takich jak: nadciśnienie wrotne, wodobrzusze, encefalopatia, dysfunkcja procesów syntezy i zaburzenia zdolności metabolicznych hepatocytów. W tym świetle stało się jasne, że wysiłki podjęte w celu wyjaśnienia mechanizmów wątrobowej fibrogenezy oraz próby zahamowania lub ograniczenia tego procesu, mają bezpośrednie implikacje kliniczne.
Aktywacja komórek gwiaździstych w procesie uszkodzenia wątroby
Patofizjologia włóknienia wątroby pozostaje w ścisłym związku z procesem aktywacji wątrobowych komórek gwiaździstych (HSC). Proces aktywacji tych komórek składa się z dwóch etapów: 1. inicjacji – zwanym również fazą przedzapalną; 2. rozwinięcia i podtrzymywania aktywacji, a kończy się fazą rezolucji w okresie, gdy choroba wątroby wygasa (3, 9, 10, 11). Efekty aktywacji HSC przedstawiono na rycinie 1.
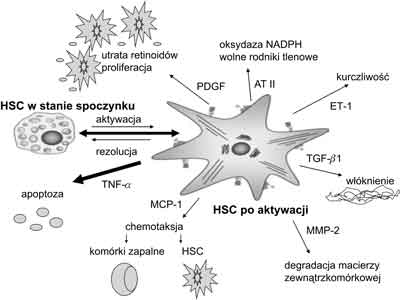 Ryc. 1. Aktywacja komórki gwiaździstej wątroby i jej efekty.
HSC – komórki gwiaździste wątroby AT II – angiotensyna II ET-1 – endotelina 1 TGF-β1 – transformujący czynnik wzrostu β1 MMP-2 – metaloproteinaza 2 MCP-1 – monocytarne białko chemotaktyczne 1 TNF-α – czynnik martwicy nowotworów α PDGF – płytkowy czynnik wzrostu Faza inicjacji obejmuje wczesne zmiany w ekspresji genów i fenotypu w odpowiedzi głównie na stymulację parakrynną płynącą praktycznie ze wszystkich rodzajów sąsiadujących komórek, w tym endotelialnych zatok, hepatocytów i płytek krwi oraz ze zmian zachodzących w otaczającej macierzy zewnątrzkomórkowej (2, 4, 9). Uszkodzone hepatocyty są bogatym źródłem wolnych rodników tlenowych wytwarzanych pod wpływem uszkodzenia błon komórkowych i peroksydacji lipidów (12, 13). Z drugiej strony, apoptoza komórek wątrobowych jako następstwo oddziaływania czynników uszkadzających stymuluje fazę inicjacji aktywacji HSC drogą białka receptorowego Fas (tzw. receptor śmierci) lub liganda indukującego apoptozę (TRIAL) zależnych od czynnika martwicy nowotworów (13). Apoptoza hepatocytów wyzwala proces włóknienia zarówno w kulturach komórkowych HSC, jak i in vivo u zwierząt doświadczalnych, a także aktywuje komórki Kupffera.
Aktywacja i infiltracja komórek Kupffera również odgrywa istotną rolę w aktywacji HSC (1, 2, 11). Wydzielają one cytokiny (głównie transformujący czynnik wzrostu β1 – TGF β1) i wolne rodniki tlenowe, które stymulują uwalnianie retinoidów, proliferację komórkową i syntezę macierzy zewnątrzkomórkowej przez HSC.
Komórki endotelium prawdopodobnie również uczestniczą w tym procesie, ponieważ poprzez konwersję latentnego TGFβ do formy aktywnej oraz syntezę komórkowej izoformy fibronektyny mogą prowadzić do wczesnej aktywacji HSC (1, 3, 4).
Płytki krwi potęgują parakrynną stymulację HSC, wydzielając płytkowy czynnik wzrostu (PDGF), TGF β1 i epidermalny czynnik wzrostu (EGF) (1, 3, 4).
Faza rozwinięcia i podtrzymywania aktywacji jest rezultatem stałej stymulacji mającej na celu zachowanie fenotypu aktywowanej HSC oraz postępującego włóknienia. Utrzymywanie aktywności HSC odbywa się na drodze zarówno autokrynnej, jak i parakrynnej. Proces aktywacji HSC obejmuje szereg dyskretnych zmian behawioralnych tych komórek, jak: utrata retinoidów, proliferacja, chemotaksja, fibrogeneza, kurczliwość oraz degradacja macierzy (1). Końcowym efektem tych zmian jest wzmożona akumulacja macierzy zewnątrzkomórkowej oraz wymiana prawidłowej macierzy na typową dla gojących się „blizn”.
Utrata retinoidów. Około 50-80% całkowitego retinolu w organizmie magazynowane jest w wątrobie, z czego 80-90% w komórkach gwiaździstych (1). Wczesnym markerem aktywacji HSC jest utrata charakterystycznych okołojądrowych kropelek witaminy A. Po wewnątrzkomórkowej hydrolizie estrów wydzielana jest ona na zewnątrz w formie retinolu (1, 9). Nie wyjaśniono do tej pory, czy zjawisko to jest przyczyną, czy skutkiem przemiany funkcjonalnej komórki.
Namnażanie komórek gwiaździstych. Najsilniejszym dotychczas zidentyfikownym mitogenem dla HSC okazał się czynnik płytkowy PDGF (1, 2). Ponadto proliferację tych komórek pobudzają inne molekuły: naczyniowy czynnik wzrostu śródbłonka naczyniowego (VEGF), trombina i jej receptory, epidermalny czynnik wzrostu (EGF), transformujący czynnik wzrostu α (TGF α), czynnik wzrostu keratynocytów oraz fibrocytarny czynnik wzrostu (bFGF – basic fibroblast growth factor) (9). Ścieżki sygnalizacyjne z udziałem wymienionych mitogenów oferują potencjalne możliwości interwencji terapeutycznej w procesie włóknienia wątroby.
Chemotaksja. HSC pobudzane przez cytokiny mogą migrować do miejsc zmienionych chorobowo. Zjawisko to częściowo tłumaczy ich obecność w przegrodach zapalnych in vivo. Zidentyfikowano kilka silnych czynników chemotaktycznych: PDGF, monocytarne białko chemotaktyczne-1 (MCP-1 – monocyte chemotactic protein-1) i CXCR3 (9). Adenozyna osłabia natomiast chemotaksję i przez to może zatrzymywać komórki w ogniskach uszkodzenia (14).
Fibrogeneza. Podstawą postępującego procesu włóknienia są dwa zjawiska: 1. proliferacja i wzrost liczby aktywnych HSC oraz 2. zwiększona synteza elementów macierzy (głównie kolagenu typu I) w pojedynczych komórkach (5, 7, 9).
Najsilniejszym stymulantem syntezy kolagenu typu I i innych komponentów macierzy (fibronektyny komórkowej, proteoglikanów) jest TGF-β1 (2, 6, 9). Odgrywa on kluczową rolę w procesie włóknienia wątroby ze względu na indukcję 3 głównych molekularnych etapów w procesie włóknienia wątroby: 1. aktywację HSC; 2. stymulację syntezy macierzy zewnątrzkomórkowej; 3. supresję degradacji macierzy zewnątrzkomórkowej (13). Rolę TGF-β1 w procesie włóknienia wątroby przedstawiono w tabeli 1.
Tabela 1. Rola transformującego czynnika wzrostu β1 w procesie włóknienia wątroby.
TGF-β1 – transformujący czynnik wzrostu β1
mRNA – matrycowy kwas rybonukleinowy TGF-β1 uwalniany przez komórki Kupffera oraz komórki endotelium zatok prowadzi do apoptozy hepatocytów, indukuje aktywację i rekrutację komórek zapalnych w ognisku uszkodzenia wątroby oraz różnicowanie komórek rezydujących w wątrobie (fibroblastów, HSC, komórek epitelium) do miofibroblastów produkujących kolagen. HSC również syntetyzują TGF-β1, a przez to pogłębiają uszkodzenie hepatocytów oraz nasilają infiltrację limfocytarną. Chociaż hepatocyty same nie produkują TGF-β1, jednak odgrywają istotną rolę w przemianie nieaktywnej formy tej cytokiny w formę aktywną biologicznie. Fibrogenne działanie TGF-β1 wynika również z faktu aktywacji przez tę molekułę fibroblastów dróg wrotnych oraz rekrutacji do stref uszkodzenia wątroby miofibroblastów z układu krążenia.
Potężnymi stymulatorami produkcji macierzy zewnątrzkomórkowej są produkty peroksydacji lipidów, a ich efekt potęguje utrata zdolności antyoksydacyjnych w aktywnych HSC (12, 13). W świetle tych doniesień zasadne wydają się badania nad możliwością wykorzystania antyoksydantów w terapii różnych chorób wątroby.
Czynnik wzrostu tkanki łącznej (CTGF/CCN2) to kolejny sygnał fibrogenny dla HSC. Jego ekspresja zwiększa się w przypadku hiperglikemii i hyperinsulinemii.
Kurczliwość. Stwierdzono, że przegrody łącznotkankowe, typowe dla schyłkowej marskości wątroby, zawierają znaczną liczbę aktywnych HSC. Kurczliwość tych komórek determinuje wzrost oporów w krążeniu wrotnym w trakcie postępującego włóknienia wątroby. Zaburzenia przepływu krwi w naczyniach wrotnych są rezultatem z jednej strony obkurczania przez HSC pojedynczych zatok, a z drugiej strony, kontrakcji marskiego narządu. Kurczliwość HSC pozostaje pod kontrolą głównie endoteliny-1 i tlenku azotu (NO), ale regulują ją również inne mediatory, np. angiotensyna II, eikozanoidy, przedsionkowy peptyd natriuretyczny, somatostatyna i tlenek węgla (CO) (3, 13). W zaktywowanych HSC wzrasta ekspresja α-SMA i innych kurczliwych filamentów, które stanowią o potencjale kurczliwości tych komórek (2).
Degradacja macierzy. W procesie włóknienia dochodzi do zaburzeń równowagi pomiędzy syntezą i degradacją macierzy zewnątrzkomórkowej. Degradacja macierzy jest więc również istotnym zjawiskiem podlegającym regulacji i wpływom terapeutycznym. Zakłócenia produkcji normalnej macierzy wątrobowej poprzez proteazy powodują akumulację macierzy bliznowatej wywierającej negatywny wpływ na funkcjonowanie komórek. Ponadto stanowią one podstawę inwazji nowotworowej i desmoplazji (3, 13). Z drugiej strony, resorpcja nadmiaru macierzy u chorych z przewlekłą chorobą wątroby daje możliwość odwrócenia dysfunkcji wątroby i nadciśnienia wrotnego. Kluczową rolę w remodelingu macierzy wątrobowej odgrywają metaloproteinazy (zwane również matriksynami). Te zależne od wapnia enzymy rozkładają kolagen i niekolagenowe substraty macierzy. Na podstawie specyficzności substratowej podzielono je na pięć klas (1, 3, 9):
1. kolagenazy miąższowe (MMP-1; MMP-8; MMP-13);
2. żelatynazy (MMP-2; MMP-9; białko aktywujące fibroblasty);
3. stromielinazy (MMP-3; MMP-7; MMP-10; MMP-11;
4. metaloproteinazy typu błonowego (MMP-14; MMP-15; MMP-16; MMP-17; MMP-24; MMP-25);
5. metaloelastaza (MMP-12).
HSC są głównym źródłem MMP-2; MMP-9; MMP-13 i stomielizyny. Istotnie podwyższoną ekspresję MMP-2 stwierdzano w marskości wątroby. Główna proteaza degradująca kolagen typu I to MMP-1. Do tej pory jednak nie udało się ustalić źródła tego enzymu. W HSC wykryto tylko niewielką jego ilość.
Aktywność metaloproteinaz podlega regulacji tkankowych inhibitorów metaloproteinaz (TIMP). Synteza TIMP-1 i TIMP-2 odbywa się w HSC, co w przebiegu uszkodzenia wątroby może hamować aktywność miąższowych kolagenaz i prowadzić do zmniejszonej degradacji, a następnie akumulacji macierzy. TIMP-1 hamuje apoptozę HSC, więc jego przedłużona ekspresja zwiększa populację aktywnych komórek gwiaździstych.
Losy komórek gwiaździstych w fazie rezolucji choroby wątroby
Kiedy choroba wątroby wygasa, dochodzi do redukcji liczby aktywnych HSC. Odbywa się to prawdopodobnie na dwóch drogach: 1. poprzez apoptozę lub 2. powrót HSC do fenotypu spoczynkowego (nieaktywnego) (ryc. 1) (3, 4, 16). To drugie zjawisko obserwowano dotychczas jedynie w kulturach komórkowych, nie zostało ono potwierdzone in vivo. Istnieje natomiast sporo dowodów na znaczenie apoptozy HSC w procesie regresji włóknienia wątroby. Niezbędny w tym względzie jest prawidłowo funkcjonujący proteasomowy system degradacji białek, inhibitory proteasomu zaburzają programowaną śmierć komórki (18). Apoptoza aktywnych HSC może być wyzwalana poprzez receptory śmierci. Stwierdzono, że induktorami apoptozy HSC są ligandy FasLICD95L, TRAIL i czynnik wzrostu nerwów (NGF). Wzrost ekspresji Fas lub receptora TNFR-1 i ich ligandów prowadzi do apoptozy drogą zależną od kaspazy 8/kaspazy 3. Natomiast nadekspresja białek proapoptotycznych, jak p53, Bax i Bcl-2, prowadzi do programowanej śmierci komórki za pośrednictwem kaspazy-9 (16, 18).
Antagonistyczne działanie wywiera sygnalizacja poprzez receptory serotoninowe.
Ostatnie badania wykazały, że aktywne komórki NK pobudzają apoptozę HSC w mechanizmie zależnym od TRAIL (3). U myszy genetycznie pozbawionych komórek NK, proces włóknienia był zdecydowanie bardziej zaawansowany niż w grupie kontrolnej (3, 13). Doniesienia te potwierdziły antyfibrotyczne działanie komórek NK.
Podobne wnioski wysnuto z obserwacji klinicznych. Nasilone włóknienie wątroby, ze względu na znacznie zaburzoną funkcję komórek NK, stwierdzano u chorych poddanych skojarzonemu leczeniu immunosupresyjnemu (cyklosporyną i kortykosteroidami) (1).
Aktywność komórek NK słabnie z wiekiem, co może częściowo wyjaśniać, dlaczego aktywność włóknienia wątroby z wiekiem narasta (1).
Poza komórkami NK w eliminacji aktywnych HSC mogą odgrywać rolę również inne komórki: wątrobowa populacja dużych, granularnych limfocytów i specyficzne dla wątroby komórki γδ T (NKT). Ich aktywność wzmaga IFN-γ (13).
Apoptoza HSC eliminuje główne źródło kolagenu oraz źródło tkankowego inhibitora metaloproteinaz (TIMP-1) – inhibitora degradacji macierzy. W rezultacie wzrasta aktywność miąższowej kolagenazy (MMP-13) syntetyzowanej przez komórki Kupffera i/lub komórki zapalne, co stanowi podstawę fazy rezolucji włóknienia (3, 9). W tabeli 2 przedstawiono poznane dotychczas induktory i inhibitory apoptozy HSC.
Tabela 2. Induktory i inhibitory apoptozy komórek gwiaździstych wątroby.
NF-κB – nuclear factor kappa-light-chain-enhancer of activated B cells – jądrowy czynnik transkrypcyjny
MAPK – mitogen activated protein kinases – kinazy białkowe aktywowane miogenem MMP-9 – metaloproteinaza 9 NGF – nerve growth factor – czynnik wzrostu nerwów IGF-1 – insulin – like growth factor – insulinowy czynnik wzrostu 1 IFN γ – interferon gamma TRAIL – TNF-related apoptosis – inducing ligand – indukujacy apoptozę ligand zależny od czynnika martwicy guzów TIMP-1 – tissue inhibitor of metalloproteinase 1 – tkankowy inhibitor metaloproteinaz 1 TGF β – transformujący czynnik wzrostu beta TNF α – czynnik martwicy guzów alfa IFN α – interferon alfa Wnioski i perspektywy na przyszłość
Obecnie nie ma wątpliwości, że aktywacja HSC pozostaje kluczowym zjawiskiem w procesie włóknienia wątroby. Publikowane wyniki kolejnych badań dostarczają dowodów na udział tych komórek również w procesach rozwoju i regeneracji wątroby, reakcjach na ksenobiotyki, w przemianach metabolicznych i immunoregulacji (zdolność prezentacji antygenów, indukcja tolerancji immunologicznej, interakcje z komórkami pochodzącymi ze szpiku kostnego) (18, 19). Szczególnie intrygujące wydają się najnowsze informacje o wyjątkowej plastyczności fenotypu i funkcji HSC oraz ich znaczeniu w namnażaniu i różnicowaniu progenitorowych komórek wątrobowych (20). Daje to potencjalną możliwość wykorzystania tych komórek do wspomagania różnicowania hepatocytów w hodowlach komórkowych i ewentualnej repopulacji in vivo. Wydaje się również, że HSC mogą zabezpieczać funkcję hepatocytów ex vivo, co z kolei stwarza nadzieję na rozwój metod zewnątrzustrojowego wspomagania pracy wątroby (1).
Podsumowując, należy stwierdzić, że HSC pomimo imponujących postępów wiedzy na ich temat, nadal pozostają ekscytującym polem do kontynuacji badań dla specjalistów różnych dziedzin nauk medycznych. Piśmiennictwo
1. Friedman SL: Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008; 88: 125-172.
2. Atzori L, Poli G, Perra A: Hepatic stellate cell: A star cell In the liver. Int J Biochem Cell Biol 2009 (article in press).
3. Friedman SL: Hepatic fibrosis-Overview. Toxicology 2008; 254: 120-129.
4. Friedman SL: Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008; 134 (6): 1655-69.
5. Henderson NC, Forbes SJ: Hepatic fibrogenesis: From within and outwith. Toxicology 2008; 254: 130-135.
6. Gressner AM, Weiskirchen R: Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β as major players and therapeutic targets. Journal of Cellular and Molecular Medicine 2006; 10: 76-99.
7. Bataller R, Brenner DA: Liver fibrosis. J Clin Invest 2005; 115: 209-18.
8. Tsukada S, Parsons CJ, Rippe RA: Mechanisms of liver fibrosis. Clin Chim Acta 2006; 364: 33-60.
9. Hui AY, Friedman SL: Molecular basis of hepatic fibrosis. Exp Rev Mol Med 2003; 5: 1-23.
10. Svegliati-Baroni G, De Minicis S, Marzioni M: Hepatic fibrogenesis in response to chronic liver injury: novel insights on the role of cell-to-cell interaction and transition. Liver International 2008; 28: 1052-1064.
11. Mortira RK: Hepatic Stellate Cells and Liver Fibrosis. Arch Pathol Lab Med 2007; 131: 1728-1734.
12. Novo E, Parola M: Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenesis and Tissue Repair 2008; 1: 5.
13. Kisseleva T, Brenner DA: Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol 2006; 21: s 84-S 87.
14. Hashmi AZ, Wyel Hakim W, Kruglov EA et al.: Adenosine inhibits cytosolic calcium signals and chemotaxis in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol 2007; 292: G 395-G 401.
15. Iredale JP: Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest 2007; 117: 539-548.
16. Elsharkawy AM, Oakley F, Mann DA: The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis 2005; 10: 927-939.
17. Watanabe A, Sohail MA, Gomes DA et al.: Inflammasome – mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol 2009; 296: G 1248-G 1257.
18. Iredale JP: Defining therapeutic targets for liver fibrosis: Exploiting the biology of inflammation and repair. Pharmacological Research 2008; 58: 129-136.
19. Winau F, Christian Quack C, Alexandre Darmoise A et al.: Starring stellate cells in liver immunology. Curr Opinion in Immunology 2008; 20: 68-74.
20. Enami Y, Bandi S, Kapoor S et al.: Hepatic stellate cells promote hepatocyte engraftment in rat liver after prostaglandin – endoperoxide synthase inhibition. Gastroenterology 2009.
otrzymano/received: 2009-10-30 zaakceptowano/accepted: 2009-12-04 Adres/address: *Beata Kasztelan-Szczerbińska Katedra i Klinika Gastroenterologii z Pracownią Endoskopową, Uniwersytet Medyczny w Lublinie ul. Jaczewskiego 8, 20-950 Lublin tel.: +48 (81) 724-45-35 e-mail: beata.szczerbinska@op.pl Artykuł Komórki gwiaździste jako główny regulator sygnalizacji międzykomórkowej w procesie włóknienia wątroby w Czytelni Medycznej Borgis. |
  Proszę kliknąć w wybraną okładkę aby przejść na stronę czasopisma
 |






Chcesz być na bieżąco? Polub nas na Facebooku: strona Wydawnictwa na Facebooku |