|
© Borgis - Postępy Nauk Medycznych 7, s. 431-440
Joanna Matyjasik, Bartłomiej Masojć, *Grzegorz Kurzawski
Analizy molekularne DNA i RNA w wykrywaniu dziedzicznych predyspozycji do nowotworów
DNA and RNA analyses in detection of genetic predisposition to cancer
Międzynarodowe Centrum Nowotworów Dziedzicznych Zakład Genetyki i Patomorfologii Pomorska Akademia Medyczna, Szczecin
Kierownik Zakładu Genetyki i Patomorfologii: prof. dr hab. med. Jan Lubiński Streszczenie
Pojawienie się w użyciu wielofunkcyjnych robotów laboratoryjnych umożliwiło wykorzystanie ich do izolacji DNA i RNA, normalizacji stężeń rozcieńczania badanych próbek, przygotowania reakcji PCR itp. Równoczesny rozwój oprogramowania i komputeryzacja sprawiły, że dzisiaj istnieje możliwość całkowitej automatyzacji procesu przechowywania, dobrze opisanych, łatwych do odnalezienie próbek i ich testowania, łącznie z transferem wyników. Obecnie czołowe firmy oferują sekwenatory umożliwiające jednoczesne sekwencjonowanie 96 próbek w oparciu o elektroforezę kapilarną produktów otrzymanych metodą cykliczną z użyciem dideoksynukleotydów znakowanych barwnikami fluorescencyjnymi. Postęp w tej dziedzinie polegał na opracowaniu nowych żeli i „chemii” umożliwiających analizę sekwencji jednego fragmentu długości prawie 1000 zasad. Oferowane nowe aparaty (GSFLX machine 2007) oparte o równoczesne sekwencjonowanie w czasie rzeczywistym bardzo wielu stosunkowo krótkich fragmentów DNA (około 200 zasad), wykorzystują pirosekwencjonowanie z detekcją luminescencji powstającej przy udziale ATP, lucyferazy lucyferyny. Aparaty te pozwalają na analizę 100 mln zasad jednego dnia. Coraz częściej stosowane są metody PCR w czasie rzeczywistym z użyciem sond TaqMana (i modyfikacji tej metody zwanej w skrócie MGB). Do wykrywania rearanżacji w genach odpowiedzialnych za dziedziczne predyspozycje do nowotworów i innych chorób stosuje się technikę MLPA, która w oparciu o reakcję ligacji specyficznych sond i reakcję amplifikacji pozwala na ocenę liczby kopii eksonów. Na jej podstawie można wnioskować o delecjach bądź duplikacjach fragmentów lub całych genów. Oferowane gotowe zestawy sond dotyczą najważniejszych znanych genów silnie predysponujących do nowotworów takich jak: ATM, BRCA1, BRCA2, CHEK1, MLH1, MSH2, MSH6, PMS2, APC, FANCA, FANCD2, PTCH, BMPR1A, SMAD4, TP53, CDH1, MEN1, NF1, NF2, STK11, SMARCB1, RB1, CDKN2A-CDKN2B, WT1. Słowa kluczowe: techniki, zmiany konstytucyjne, diagnostyka
Summary
Introduction of liquid handling robots made possible their application in DNA or RNA isolation, normalization of samples´ concentration, PCR preparation, etc. Parallel development of software and hardware enabled complete automatic management of large sample series in genetic testing, including data transfer without any user intervention. Nowadays leading companies offer capillary sequencers analyzing simultaneously up to 96 samples amplified by means of cycling sequencing with fluorescent dyes. Improved "chemistry” and gels´ composition enabled accurate sequencing of 1000 bp fragments. Another system (GSFLX machine 2007), based on massively parallel sequencing by synthesis technology can generate 100 Mb of genomic sequence in a single run (about 4.5 h). Real-time PCR techniques with TaqMan(r) probes (or TaqMan(r) MGB probes) have become commonly used in laboratory practice. MLPA technique used in detection of rearrangements in genes associated with hereditary cancers allows the determination of exon copy number. The presence of deletions or duplications of exons or whole genes can be analyzed by that method. Commercial kits are available for genes with a well-documented association with hereditary cancers: ATM, BRCA1, BRCA2, CHECK1, MLH1, MSH2, MSH6, PMS2, APC, FANCA, FANCD2, PTCH, BMPR1A, SMAD4, TP53, RB1, CDKN2A - CDKN2B, WT1, CDH1, MEN1, NF1, NF2, STK11, SMARCB1. Key words: techniques, constitutional changes, diagnoses
W ostatnich latach zidentyfikowano szereg genów, których mutacje odpowiedzialne są za wysoką dziedziczną predyspozycję do nowotworów (1).
U nosicieli mutacji tych genów ryzyko zachorowania na chorobę nowotworową może wynosić nawet 90%. Wybrane geny związane z wysoką genetyczną predyspozycją do nowotworów i najczęściej badane w praktyce lekarskiej zestawiono w tabeli 1.
Tabela 1. Geny, których mutacje predysponują do zespołów nowotworów dziedzicznych. Zestawienie obejmuje geny najczęściej badane w naszym laboratorium.
Opracowano szereg metod molekularnych, które pozwalają na wykrywanie mutacji. Można je podzielić na metody wykrywania:
I) nieznanych mutacji,
II) znanych mutacji/polimorfizmów.
I. Wykrywanie nieznanych mutacji
Zastosowanie technik wchodzących w skład tych metod w odpowiednio dobranych pod względem cech rodowodowo-klinicznych przypadkach jest w praktyce lekarskiej uzasadnione mimo tego, że techniki te jak dotychczas są złożone, pracochłonne i kosztowne.
Techniki bezpośredniego wykrywania nosicielstwa mutacji
Analizy DNA
Zasadnicze rodzaje analiz:
a) izolacja DNA
b) amplifikacja fragmentów genów, z reguły sekwencji kodujących
c) wstępne wykrywanie zaburzeń w produktach amplifikacji technikami przesiewowymi
d) sekwencjonowanie
e) metoda Southerna i zależna od ligacji multipleksowa amplifikacja sond.
Izolacja DNA
Materiał do izolacji DNA stanowią na ogół komórki łatwo dostępne takie jak leukocyty z krwi obwodowej lub rzadziej bioptaty innych tkanek. W trakcie analiz wykrywana jest mutacja konstytucyjna, a więc obecna we wszystkich komórkach pacjenta. Materiał do badania najlepiej pobrać bezpośrednio przed izolacją, ale dobre wyniki uzyskuje się również po kilkudniowym przechowywaniu krwi w temperaturze pokojowej lub nawet przez kilka lat w temperaturze poniżej zera. Jeżeli nie dysponujemy tkankami świeżymi to izolację DNA można wykonać z tkanek utrwalonych w formalinie i zatopionych w bloczkach parafinowych, chociaż uzyskanie jednoznacznych wyników z takiego materiału jest trudne, niekiedy wręcz niemożliwe. Izolowanie DNA polega na usunięciu białek z lizatu komórkowego. W metodzie fenolowo-chloroformowej uzyskuje się to poprzez trawienie proteinazą K i ekstrakcję w mieszaninie fenolu i chloroformu. Z odbiałczonych w ten sposób próbek kwasy nukleinowe wytrąca się alkoholami: etylowym lub izopropylowym. Mało kto dzisiaj w codziennej pracy używa tej czasochłonnej i toksycznej, choć dającej czyste i nie zdegradowane DNA metody (jest ona nadal stosowana jednynie do izolacji DNA z „bloczków parafinowych”). Zastąpiły ją inne mniej pracochłonne metody, które łatwiej poddają się procesowi automatyzacji, a są oparte na wybiórczym wiązaniu DNA z nośnikiem (złoże chromatograficzne, filtr bądź kuleczki magnetyczne) następnie odmyciu zanieczyszczeń i uwolnieniu DNA do roztworu.
Amplifikacja fragmentów genów
W tej analizie powielane są fragmenty badanego DNA za pomocą reakcji łańcuchowej polimeryzacji(PCR). W skład mieszaniny reakcyjnej wchodzą: matryca DNA (zwykle genomowe DNA), polimeraza DNA, para specyficznych starterów („primers”), trójfosforany deoksyrybonukleotydów oraz bufor reakcyjny. Mieszanina ta poddawana jest w specjalnym termostacie cyklicznym zmianom temperatury. Każdy cykl składa się z trzech etapów: denaturacji, przyłączania starterów i syntezy. Po 22 cyklach, przy 100% wydajności, liczba kopii powielanego fragmentu zwiększa się milion razy.
Wstępne wykrywanie zaburzeń w produktach amplifikacji technikami przesiewowymi
Niegdyś popularną techniką wstępnego wykrywania zaburzeń w produktach amplifikacji było badanie zmian konformacji jednoniciowego DNA – SSCP (single stranded conformational polymorphism) (2).
Inne techniki tego rodzaju to analiza heterodupleksów – HET (heteroduplex analysis) (3), chemiczne rozszczepianie niesparowań heterodupleksów – CMC (chemical mismatch cleavage) (4), DHPLC(Denaturing High-Performance Liquid Chromatography) (5) i elektroforeza na żelach z gradientem czynnika denaturującego – DGGE (denaturing gradient gel electrophoresis) (6).
DHPLC – wysokosprawna denaturująca chromatografia cieczowa
Najlepszą i najczęściej używaną techniką wstępnego wykrywania zmian jest obecnie DHPLC (5, 7, 8, 9, 10). Jest to odmiana HET wykorzystująca wysoką rozdzielczość nowoczesnych wypełnień kolumn chromatograficznych. Rozdział analizowanych fragmentów DNA przeprowadzany jest w gradiencie czynnika denaturującego. W warunkach subdenaturacyjnych heterodupleksy wykazują mniejsze powinowactwo niż homodupleksy do złoża kolumny i łatwiej ulegają wymyciu. Całość rozdziału monitorowana jest przez miernik absorbancji mierzonej przy 260 nm. Profil elucji (ryc. 1) jest charakterystyczny i powtarzalny dla danej zmiany i pozwala na odróżnienie nowych zmian od wcześniej wykrytych mutacji bądź polimorfizmów.
 Ryc. 1. Profil elucji DHPLC charakterystyczny (przerywana linia) dla mutacji A na G w eksonie 19 genu MLH1.
Z danych literaturowych (11) i badań własnych (12) wynika, że DHPLC łączy zalety dotychczas stosowanych metod. Czułość metody sięga 100% (5, 9, 10) przy stosunkowo niskich kosztach (koszt odczynników najwyżej 20 złotych na próbkę) jest ona szybka, a przy zastosowaniu „autosamplera” pozwala wykonać analizę 200 próbek na dobę.
Sekwencjonowanie
Sekwencjonowanie jest najbardziej czułą techniką wykrywania zmian w materiale genetycznym umożliwiającą jednocześnie ich pełną charakterystykę. W ostatnich latach znaczny postęp w technologii sekwencjonowania osiągnięto poprzez wprowadzenie automatycznych aparatów do sekwencjonowania, których funkcjonowanie oparte jest o fluorescencję wzbudzaną laserem. Każdy z nukleotydów (A, C, G, T) może być wyznakowany innym fluorochromem (ryc. 2). Najbardziej dogodną techniką jest sekwencjonowanie metodą cykliczną (13). W trakcie badania oceniane są sekwencje produktów PCR obu nici DNA. Rzeczywista zmiana w odróżnieniu od artefaktów wykrywana jest w obu niciach (ryc. 3). Procedura sekwencjonowania składa się z kilku etapów:
 Ryc. 2. Obraz żelu sekwencyjnego – wynik rozdziału asymetrycznego PCR (nukleotyd adeninowy wyznakowany fluorochromem zielonym; tymidynowy żółtym; guanidynowy niebieskim; cytozynowy czerwonym).
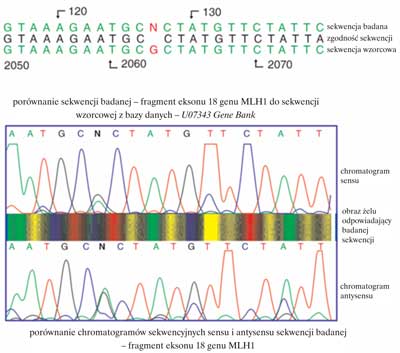 Ryc. 3. Wstępna analiza sekwencji z wykrytą zmianą w obrębie eksonu 18 genu MLH1.
1) preparatywnego PCR – polegającego na namnożeniu wybranego fragmentu genu przy użyciu pary specyficznych starterów;
2) asymetrycznego PCR – dla każdej próbki amplifikacja osobno z każdym ze starterów z zastosowaniem dideoksynukleotydów znakowanych barwnikami fluorescencyjnymi;
3) elektroforezy na denaturującym żelu poliakrylamidowym z równoczesną detekcją i rejestracją przepływających produktów;
4) analizy otrzymanych wyników przy użyciu pakietu programów komputerowych.
Podczas asymetrycznego PCR powstają wszystkie możliwe, różniące się długością oligonukleotydy komplementarne do matrycy i zawierające na 3´-końcu fluorochrom („zaznaczone kolorem”). Zostają one rozdzielone podczas elektroforezy od najkrótszego po najdłuższy, a kolejność kolorowych nukleotydów odczytana jako sekwencja komplementarna do matrycy. Stwierdzana sekwencja DNA jest później porównywana z sekwencją prawidłową z dostępnych baz danych takich jak GenBank i EMBL. Przez porównanie z sekwencjami prawidłowymi określany jest dokładnie charakter zmiany (ryc. 3 i 4).
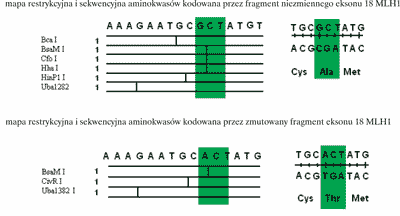 Ryc. 4. Zmiany mapy restrykcyjnej i sekwencji aminokwasów w białku spowodowane mutacją w eksonie 18 genu MLH1. Mutacja prowadzi do utraty miejsc restrykcyjnych dla Bca I, Cfo I, Hha I, HinP I oraz pojawienia się miejsca restrykcyjnego dla Cvi R I, co łatwo można wykorzystać do jej wykrywania.
Obecnie czołowe firmy oferują sekwenatory umożliwiające jednoczesne sekwencjonowanie 96 próbek w oparciu o elektroforezę kapilarną produktów otrzymanych metodą cykliczną z użyciem dideoksynukleotydów znakowanych barwnikami fluorescencyjnymi. Postęp w tej dziedzinie polegał w ostatnich latach nie tylko na zwiększeniu liczby jednocześnie analizowanych próbek, ale na opracowaniu nowych żeli (umożliwiających wielokrotny rozdział na tym samym wypełnieniu kapilary) i „chemii” (mieszaniny złożonej z buforów; substratów, polimerazy i tak zwanych ulepszaczy) umożliwiających analizę sekwencji jednego fragmentu długości prawie 1000 zasad.
Oferowane są też nowe aparaty GSFLX machine 2007oparte o równoczesne sekwencjonowanie w czasie rzeczywistym przez syntezę równoczesną bardzo wielu stosunkowo krótkich fragmentów DNA (około 200 zasad). Wykorzystują one pirosekwencjonowanie z detekcją luminescencji powstającej z rozkładu ATP. Aparaty te pozwalają na analizę 100 mln zasad jednego dnia. Pirosekwencjonowanie (14) jako matrycę wykorzystuje jednoniciowy fragment DNA, na którym przeprowadzana jest synteza nici komplementarnej poprzez dodawanie kolejno czterech różnych trifosforanów deoksynukleotydów (dNTP). Przyłączeniu każdej zasady towarzyszy uwalnianie pirofosforanu, który zostaje przekształcony w ATP przy udziale sulfurylazy i obecnego w mieszaninie adenozyno-5´fosfosiarczanu. Powstały ATP wykorzystywany jest przez lucyferazę do przekształcenia lucyferyny w oksylucyferynę. W reakcji tej powstaje światło w ilości odpowiadającej wyprodukowanemu we wcześniejszym etapie pirofosforanowi. Światło to jest rejestrowane przez kamerę CCD i przekształcane do postaci piku na wykresie. Ten sam schemat reakcji przeprowadzany jest również dla kolejno dodawanych różnych dNTPów. Jeżeli dodawany nukleotyd nie jest komplementarny do matrycy nie następuje włączenie go do nowo syntetyzowanej nici i nie powstaje pirofosforan. Tylko obecność sygnału świetlnego stanowi podstawę do zapisu w sekwencji kolejnego dodawanego nukleotydu.
Mutacje w DNA obejmujące miejsca przyłączania starterów lub inne odcinki poza regionami amplifikowanymi nie są wykrywane za pomocą testów DNA opartych o analizy omówione powyżej. Część takich zmian to duże przemieszczenia.
Metoda Southerna i zależna od ligacji multitpleksowa amplifikacja sond
Jedną z technik, kiedyś powszechnie używaną do wykrywanie dużych przemieszczeń opartą o hybrydyzację jest opisana po raz pierwszy przez E.M. Southerna w 1975 r. i odtąd zwana metodą Southerna (Southern bloting).
Współcześnie metodą godną polecenia, która niemal całkowicie zastąpiła metodę Southerna w wykrywania rearanżacji w genach jest zależna od ligacji multitpleksowa amplifikacja sond ( MLPA – multiplex ligation-dependent probe amplification) (15). Technika ta w oparciu o reakcję ligacji specyficznych sond i reakcję amplifikacji pozwala na ocenę liczby kopii eksonów. Na jej podstawie można wnioskować o delecjach bądź duplikacjach fragmentów lub całych genów (geny odniesienia jako kontrola). W technice tej stosuje się wiele par sond. Sondy zawierają oprócz sekwencji docelowych komplementarnych do sekwencji eksonowych (sekwencje ulegające hybrydyzacji), sekwencje starterowe, a jedna z każdej pary dodatkowo unikatową sekwencję wstawki (ryc. 5).
 Ryc. 5. Budowa sond w MLPA.
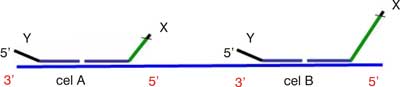
Sekwencje hybrydyzujące każdej pary sond wiążą się komplementarnie do sąsiadujących ze sobą fragmentów DNA i tylko przy w pełni komplementarnej hybrydyzacji może zajść ligacja (ryc. 6).
 Ryc. 6. Hybrydyzacja i ligacja sond.
Po przyłączeniu sond do matrycy następuje ich ligacja, a następnie denaturacja. Oddysocjowana zligowana sonda zawierająca sekwencje startowe zostaje poddana amplifikacji w reakcji PCR. Obecność różnej długości wstawek pozwala na rozróżnienie produktów skierowanych na różne cele, a ilość produktu jest proporcjonalna do ilości sekwencji matrycowej. Każdy pik odpowiada produktowi amplifikacji zligowanej specyficznej pary sond (ryc. 7).
 Ryc. 7. Wynik elektroforezy próby kontrolnej.
Różnice względne w wysokości bądź powierzchni piku wskazują na zmiany ilościowe (bądź czasami jakościowe) docelowej sekwencji sondy (ryc. 8).
 Ryc. 8. Wynik elektroforezy próby badanej wskazujący na delecję eksonu 13.
Zaletą tej techniki jest niewielka ilość DNA wymagana do przeprowadzenia analizy i możliwość zastosowania nawet zdegradowanego materiału genetycznego. Najważniejszymi zaletami metody są prostota wykonania, niska cena i małe wymagania co do ilości (20 ng genomowego DNA) i jakości DNA. Oferowane gotowe zestawy sond dotyczą najważniejszych znanych genów silnie predysponujących do nowotworów takich jak: ATM, BRCA1, BRCA2, CHEK1, MLH1, MSH2, MSH6, PMS2, APC, FANCA, FANCD2, PTCH, BMPR1A, SMAD4, TP53, CDH1, MEN1, NF1, NF2, STK11, SMARCB1, RB1,CDKN2A-CDKN2B, WT1.
Analizy RNA
Zalety analiz RNA wynikają przede wszystkim z możliwości wykrycia mutacji przy wykonaniu mniejszej liczby reakcji (co wynika z mniejszej długości RNA określonej liczbą zasad w porównaniu do DNA). Jak dotychczas główne wady tych technik obejmują trudności w uzyskiwaniu powtarzalnych wyników, mniejszą stabilność RNA z niektórymi mutacjami oraz kłopoty w interpretacji związane z występowaniem różnych form składania RNA ( alternative splicing).
Etapy badania obejmują:
a) izolację RNA
b) amplifikację części kodujących genów
c) wykrywanie zaburzeń w produktach amplifikacji.
Izolacja RNA
W większości pracowni RNA izolowane jest z limfocytów krwi obwodowej. Izolację RNA przeprowadza się w podobny sposób jak izolację DNA. Ze względu na wszechobecność termostabilnych RNaz w tkankach, izolacja RNA wymaga większej staranności. Powszechnie stosowaną metodą izolacji jest procedura P. Chomczyńskiego (16) polegająca na lizie komórek w roztworze rodanku guanidyny (inhibitor RNaz) i następnie ekstrakcji mieszaniną fenolu i chloroformu. Lekko kwaśny odczyn fenolu sprawia, że wytrąceniu oprócz białek ulega również DNA, który w tych warunkach jest praktycznie nierozpuszczalny.
RT/PCR – Odwrotna transkrypcja z reakcją PCR (reverse transcription PCR)
RNA można przepisywać na cDNA (komplementarne DNA) za pomocą odwrotnej transkryptazy i namnożyć przy pomocy reakcji PCR. RNA nie zawiera intronów, więc do powielenia kodującej części wybranego genu wystarcza zwykle zaledwie kilka par starterów. Oceniając tak otrzymane cDNA na zwykłych żelach agarozowych wykryć można zaburzenia RNA polegające na delecjach lub insercjach fragmentów o długości powyżej kilkudziesięciu par zasad.
Wykrywanie zaburzeń w produktach amplifikacji
Produkt reakcji RT/PCR może być też badany wszystkimi wcześniej omówionymi technikami oraz przy pomocy testu syntezy białka in vitro – IVTT (In Vitro Transcription Translation Assay) zwanego również PTT (Protein Truncation Test) (17, 18). RT-PCR jest pierwszym etapem w PTT. W PTT jeden starter zawiera nie tylko sekwencje inicjujące przepisywanie na cDNA, ale dodatkowo sekwencje inicjujące translację tj. syntezę in vitro białka na bazie cDNA. Po rozdziale elektroforetycznym i przeniesieniu na błonę nitrocelulozową oceniana jest długość zsyntetyzowanego białka, która jest zmieniona nie tylko wówczas, gdy w obrębie RNA występują duże delecje lub insercje, ale również w przypadkach mutacji nawet pojedynczych nukleotydów prowadzących do powstania sekwencji typu stop kodon (TGA, TAA lub TAG) lub mutacji „splicingowych”. Wadą PTT jest niemożność wykrycia mutacji typu zmiany sensu. Szacuje się, że nawet przy wykorzystywaniu wszystkich znanych testów czułość bezpośredniego wykrywania zmian wynosi obecnie około 70-80%, głównie ze względu na niemożność rozpoznania zaburzeń w nieznanych sekwencjach regulujących funkcjonowanie genów.
II. Wykrywanie znanych mutacji
Coraz więcej wiadomo o rodzaju i częstości mutacji predysponujących do nowotworów charakterystycznych dla różnych populacji, w tym o mutacjach powtarzalnych, czyli występujących w wielu rodzinach danej grupy etnicznej. Testy DNA dotyczące wykrywania znanych mutacji nabierają coraz większego znaczenia ze względu na ich niezwykle wysoką efektywność ekonomiczną. Takie geny jak BRCA1, MLH1 i MSH2 czy VHL doczekały się w Polsce opracowań populacyjnych (epidemiologicznych), dzięki którym wiadomo jakich mutacji i w których miejscach genów szukać (19, 20, 21). Najczęściej stosowane testy DNA wykrywające znane mutacje wykorzystują techniki:
RFLP/PCR – ( restriction fragment-length polymorphism/PCR) – wykrywanie mutacji za pomocą enzymów restrykcyjnych w produktach łańcuchowej reakcji polimeryzacji. Enzymy restrykcyjne, zwane endonukleazami restrykcyjnymi lub krótko restryktazami rozpoznają specyficzne sekwencje zasad w dwuniciowym DNA i rozcinają obie nici dokładnie w określonym miejscu. Dlatego są wykorzystywane do wykrywania mutacji punktowych, małych delecji lub insercji, które prowadzą do utraty lub pojawienia się nowych miejsc restrykcyjnych. Namnożony produkt PCR zawierający zmianę poddaje się trawieniu odpowiednią restryktazą, a następnie rozdziela przy pomocy elektroforezy na żelu agarozowym (przykład – ryc. 9) lub poliakrylamidowym.
 Ryc. 9. Zasada RLFP/PCR. Schematyczny obraz żelu po elektroforezie – tylko wariant genu zwierający G jako zasadę produktu PCR podlega trawieniu przez restryktazę Cfo I w odróżnieniu od formy zmutowanej zawierającej w tym miejscu A.
ASA – (allele specific amplification) – wykrywanie mutacji przy pomocy specyficznych oligonukleotydów. W popularnie używanej wersji tej techniki z użyciem elektroforezy agarozowej oprócz starterów flankujących stosuje się starter w pełni komplementarny do allela z mutacją lub startery, z których jeden jest w pełni komplementarny do allela z mutacją, a drugi do allela niezmienionego. Przy tym startery są tak zlokalizowane, że w wyniku PCR powstają różne produkty (różniące się długością) w zależności od genotypu użytej próbki DNA (ryc. 10). Nowoczesna wersja tej metody wykorzystująca krótkie sondy fluorescencyjne allelospecyficzne i aparaty „ real time PCR” (22, 23) pozwala na bardzo szybkie badanie wielu próbek DNA. Technologię matryc (macierzy) z unieruchomionymi na stałej fazie oligonukleotydami można traktować jako współczesną wersję ASA. Niewątpliwą zaletą tej technologii jest daleko idąca automatyzacja i możliwość równoczesnego badania nawet kilku tysięcy znanych mutacji. Dostęp do tej technologii w polskich realiach jest bardzo ograniczony z powodu jej wysokiej ceny.
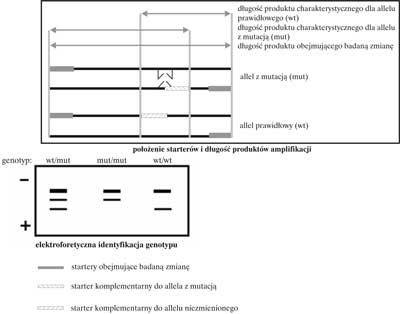 Ryc. 10. Zasada ASA.
PCR w czasie rzeczywistym ( Real Time PCR)
Jedną z najnowszych i coraz częściej stosowanych technik w biologii molekularnej jest „Real – Time PCR” pozwalający na monitorowanie ilości produktu reakcji PCR w każdym jej cyklu. Modyfikacja tej techniki polegająca na zastosowaniu fluorescencyjnie znakowanych sond komplementarnych do sekwencji badanego fragmentu DNA znalazła swoje zastosowanie również w identyfikacji znanych zmian genetycznych. Istnieje szereg systemów opartych na tej technice różniących się typem sondy zastosowanym w celu detekcji badanej zmiany. Wśród nich wyróżnia się systemy wykorzystujące między innymi sondy typu: TaqMan i SimpleProbes.
Sondy TaqMan
Stosowana w tym systemie sonda specyficzna dla amplifikowanego fragmentu znakowana jest na 5´ końcu barwnikiem reporterowym: FAM (6-karboksyfluoresceina), HEX (heksachloro-6-karboksyfluoresceina), TET (tetrachloro-6-karboksyfluoresceina) lub JOE (2,7-dimetylo-4,5-dichloro-6-6-karboksyfluoresceina) a na końcu 3´ barwnikiem tłumiącym: TAMRA (6-karboksy-tertrametylorodamina) lub DABCYL (kwas 4-(4´-dimetyloaminfenylazo)-benzoesowy). Bliskość barwnika reporterowego w stosunku do barwnika tłumiącego w obrębie tej samej sondy powoduje, iż fluorescencja jest wygaszana. Podczas reakcji PCR na etapie przyłączania starterów wyznakowana sonda wiąże się specyficznie z matrycą pomiędzy miejscami hybrydyzacji starterów. Jej 3´ koniec jest zablokowany co powoduje, iż przy następnym etapie jakim jest wydłużanie starterów nie może być ona wydłużana tak jak startery. Zastosowana w tym systemie polimeraza o aktywności 5´-3´ dobudowując nić DNA degraduje sondę, co skutkuje uwolnieniem barwnika reporterowego od barwnika tłumiącego i prowadzi do wzrostu fluorescencji. Proces ten zachodzi podczas każdego cyklu powodując narastanie sygnału fluorescencyjnego z poszczególnych cykli, co umożliwia detekcję sygnału w każdym momencie trwania reakcji. Sondy stosowane w tym systemie mają długość od 20 do 40 nukleotydów, liczba par G+C w ich sekwencji zawiera się w przedziale od 40-60%. Sondy nie powinny zawierać powtórzeń pojedynczych nukleotydów szczególnie guaniny. Sekwencja sondy nie powinna być też komplementarna do sekwencji starterów jak i do sekwencji matrycy w miejscu przyłączenia starterów. Ważne jest, aby sonda nie posiadała na 5´-końcu zasady G, ponieważ jej obecność wygasza fluorescencję barwnika reporterowego nawet po odseparowaniu go od barwnika tłumiącego.
Modyfikacją tego systemu jest zastosowanie sondy TaqMan typu MGB ( Minor Groove Binder), w której do końca 3´ przyłączona jest również grupa MGB. Jej funkcja polega na stabilizacji przyłączenia sondy poprzez wpasowanie się do kompleksu powstałego z sondy i matrycowego DNA. Interakcja grupy MGB z kompleksem sonda-matryca podnosi temperaturę topnienia sondy o 15-30°C, co pozwala na zastosowanie sond o znacznie krótszej sekwencji (od 14 do 18 nukleotydów). Jest to korzystne podczas analizy polimorfizmów pojedynczego nukleotydu, ponieważ krótkie sondy łatwiej ulegają destabilizacji pod wpływem zmian nukleotydów występujących w badanej sekwencji.
Simple Probes (Guanine quenching probes)
Guanina wykazuje cechy cząsteczki tłumiącej fluorescencję takich barwników jak FAM czy JOE. W technice tej wykorzystuje się krótki fragment jednoniciowego DNA o długości ok. 20-30 nukleotydów (sonda molekularna) o sekwencji komplementarnej do badanego DNA zawierającego zmianę/mutację, wyznakowany na 5´ lub 3´ końcu barwnikiem fluorescencyjnym (FAM lub JOE). Technika ta umożliwia zidentyfikowanie heterozygotycznych oraz homozygotycznych wariantów zmiany/mutacji poprzez pomiar wzrostu fluorescencji wykonywany w gradiencie temperatury. Sonda, która hybrydyzuje z sekwencją badaną, ma zwykle wyższą temperaturę topnienia, jeżeli hybrydyzuje z nicią w pełni komplementarną, natomiast niższą temperaturę topnienia, gdy pod sondą znajduje się pojedynczy niesparowany nukleotyd. Odczyt poziomu fluorescencji podczas podnoszenia temperatury w zakresie 40°-80°C pozwala na zidentyfikowanie konkretnego wariantu badanej zmiany w DNA.
Systemy oparte na zastosowaniu komplementarnych wyznakowanych fluorescencyjnie sond posiadają szereg zalet. Są nimi: wysoka czułość, krótki czas i pełne zautomatyzowanie analizy oraz zniwelowanie ryzyka kontaminacji poprzez przeprowadzenie wszystkich etapów w zamkniętych dołkach płytki. Wadą techniki jest konieczność projektowania sond dla poszczególnych sekwencji, jak i zbyt mała uniwersalność warunków doświadczalnych.
MALDI -TOF ( Matrix Assisted Laser Desorption/Ionization Time of Flight)
MALDI-TOF jest jedną z technik spektrofotometrii masowej, która wykorzystywana jest również w detekcji zmian w obrębie badanego fragmentu DNA. Najczęściej stosuje się ją do analizy polimorfizmów pojedynczych nukleotydów (SNP). Analizę poprzedzają etapy oparte o reakcje PCR. Pierwszy prowadzący do namnożenia wybranego bądź wybranych fragmentów (w wersji multipleksowej) zawierających badane SNP-y. Drugi asymetryczny (jeden primer komplementarny do sekwencji poprzedzającej polimorficzne miejsce) podobnie jak w sekwencjonowaniu z zastosowaniem dideoksynuklotydów. Później po oczyszczeniu za pomocą żywicy jonowymiennej próbki nanoszone są na płytki ( spektroCHIP), a następnie poddawane impulsowi laserowemu wzbudzającemu jony. Cała analiza przeprowadzana jest w warunkach próżniowych, przez co ruch jonów nie jest zakłócany przez zderzenia z cząsteczkami gazów. Prędkość przemieszczania się wzbudzonych jonów pochodzących od badanych próbek analizowana jest przez detektor czasu przelotu jonów. Jony pochodzące od większej masy docierają do detektora wolniej niż jony pochodzące od mniejszej masy. Różnice nukleotydowe występujące w badanych próbkach wpływają na ich masę, co pozwala na ich rozróżnienie. Rozdział analizowanych cząsteczek dokonywany jest na podstawie stosunku masy jonów do ich ładunku. Technika ta charakteryzuje się wysoką czułością i powala na szybkie przeprowadzenie analizy. System Sequenome MassARRAY (www.sequenom.com) oparty o MALDI-TOF pozwala na genotypowanie 76 tysięcy punktów w ciągu dnia. Tylko wysokie koszty aparatu powodują niską popularność tej metody.
Podsumowanie
W niniejszym opracowaniu przedstawiono podstawowe testy DNA i RNA stosowane w wykrywaniu mutacji konstytucyjnych u osób z genetyczną predyspozycją do nowotworów. W ciagu ostatnich lat rozwój i upowszechnianie nowych metod molekularnych przebiega bardzo szybko. Pojawienie się w użyciu wielofunkcyjnych robotów laboratoryjnych umożliwia wykorzystanie ich nie tylko do izolacji DNA i RNA, ale też do normalizacji stężeń (doprowadzenie do żądanego i jednakowego stężenia serii próbek), przygotowania reakcji PCR i itp. Równoczesny rozwój oprogramowania i komputeryzacja sprawiły, że dzisiaj istnieje możliwość całkowitej automatyzacji procesu bankowania próbek i ich testowania, łącznie z transferem danych i wyników. W laboratoriach używamy coraz mniej oddzielnych probówek. Na trwałe do użytku weszły płytki o 96 czy nawet 384 dołkach, bo większość analiz wykonujemy w dużych seriach stosując coraz mniejsze objętości odczynników (miniaturyzacja), używając automatycznych dozowników, co przekłada się na coraz mniejsze koszty analizy jednej próbki. Do lamusa historii odeszły jeszcze tak nie dawno bardzo popularne metody wykrywania nieznanych mutacji takie jak SSCP, czy metoda DGGE. Natomiast na dobre zadomowiła się w naszych laboratoriach DHPLC stając się obowiązującym standardem we wstępnym wykrywaniu mutacji. Wypieranie niektórych mniej czułych czy bardziej złożonych albo toksycznych metod jest też spowodowane znacznym postępem i zwiększeniem dostępności sekwencjonowania. W przypadku analizy długich fragmentów (około 1000 zasad) bezpośrednie sekwencjonowanie jest dziś najtańszą, najszybszą i najpewniejszą metodą wykrywania nieznanych mutacji. Obecnie czołowe firmy oferują sekwenatory umożliwiające jednoczesne sekwencjonowanie 96 próbek w oparciu o elektroforezę kapilarną produktów otrzymanych metodą cykliczną z użyciem dideoksynukleotydów znakowanych barwnikami fluorescencyjnymi. A nowe aparaty GSFLX machine 2007 pozwalają na analizę 100 mln zasad jednego dnia i pewnie już niedługo znajdą zastosowanie do szybkiego sekwencjonowania indywidualnego genomu ludzkiego bądź wielu jego rejonów odpowiedzialnych za zwiększone predyspozycje do chorób, w tym również nowotworowych. Próbie czasu oparła się ASA w wersji z użyciem elektroforezy agarozowej jednak jest coraz rzadziej używana. Z trudem broni swojej pozycji RFLP/PCR ze względu na upowszechnienie mniej pracochłonnej i tańszej metody PCR w czasie rzeczywistym zwłaszcza z użyciem sond TaqMana, która pozwala na znacznie szybsze analizowanie znanych SNP-ów i mutacji w wielu próbkach równocześnie (płytki na 384 próbek). Dodatkową zaletą tej techniki jest wyeliminowanie możliwości kontaminacji laboratorium produktami reakcji PCR, co jest niezwykle ważne zwłaszcza w laboratoriach diagnostycznych. Dzisiaj konkurecję dla tej metody stanowi niezwykle wydajny system Sequenome MassARRAY pozwalający na genotypowanie wielu tysięcy punktów polimorficznych w ciągu dnia. Niemal całkowicie z użycia wyszła żmudna i pracochłonna metoda Southerna. W wykrywaniu rearanżacji w genach odpowiedzialnych za dziedziczne predyspozycje do nowotworów i innych chorób zastąpiła ją MLPA. Piśmiennictwo
1. Lubiński J, et al.: Molecular basis of inherited predispositions for tumors. Acta Biochim Pol 2002, 49: 571-81.
2. Orita M, et al.: Detection of polimorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natn Acad Sci USA 1989, 86: 2766-70.
3. Nagamine CM, Chan K, Lau YFCA: PCR artifact: Generation of heteroduplexes. Am J Hum Genet 1989, 45: 337-9.
4. Cotton RGH, Rodrigues NR, Camphbell RD: Reactivity of cytosine and thymine in single-base-pair mismatches with hydroxylamine and osmium tetroxide and its application to the study of mutations. Proc Nat Acad Sci USA 1988, 85: 4397-401.
5. O´Donovan MC, Oefner PJ: Blind analysis of denaturing high-performance liquid chromatography as a tool for mutation detection. Genomics 1998, 52: 44-9.
6. Myers RM, Maniatis T, Lerman LS: Detection and localization of single base changes by denaturing gradient gel electrophoresis. Meth Enzymol 1987, 155: 501-27.
7. Liu W, Smith DI: DHPLC used in the detection of germline and somatic mutations. Nucleic Acids Res 1998, 26: 1396-400.
8. Jones AC, Austin J: Optimal temperature selection for mutation detection by denaturing HPLC and comparison to single-stranded conformation polymorphism and heteroduplex analysis. Clin Chem 1999, 45: 1133-40.
9. Arnold N, Gross E: A highly sensitive, fast and economical technique for mutation analysis in hereditary breast and ovarian cancers. Hum Mutat 1999, 14: 333-9.
10. Gross E., Arnold N: A comparison of BRCA1mutations analysis by direct sequencing, SSCP and DHPLC. Hum Genet 1999, 105: 72-8.
11. Xiao W, Oefner PJ: Denaturing high-performance liquid chromatography: a review. Hum Mutat 2001, 17: 439-74.
12. Kurzawski G, et al.: Mutation analysis of MLH1and MSH2genes performed by denaturing high-performance liquid chromatography. J Biochem Biophys Meth 2002, 51: 89-100.
13. Rosenthal A, Charnock JDS: New protocols for sequencing with dye terminators. DNA Seq 1992, 3: 61-4.
14. Ronaghi M, et al.:Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem 1996, 242: 84-90.
15. Schouten JP, et al.: Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002, 30: E57.
16. Chomczyński P, Sacchi N: Single-step method of RNA isolation by acid guanidinum thiocyanate-phenol-chloroform extraction. Anal Biochem 1987, 162: 156-9.
17. Luce MC, et al.: In Vitro Transcription/Translation Assay for the Screening of hMLH1and hMSH2Mutations in Familial Colon Cancer. Gastroenterology 1995, 109: 1368-74.
18. Plumer SJ, Casey G: Are we closer to genetic testing for common malignances. Nature Medicine 1996, 2: 156-8.
19. Kurzawski G, et al.: Germline MSH2and MLH1mutational spectrum in HNPCC families from Poland and the Baltic States. J Med Genet. 2002, 39, E65.
20. Cybulski C, et al.: Germline mutations in the von Hippel-Lindau (VHL) gene in patients from Poland: disease presentation in patients with deletions of the entire VHL gene. J Med Genet. 2002, 39: E38.
21. Górski B, et al.: Founder mutations in the BRCA1gene in Polish families with breast-ovarian cancer. Am J Hum Genet 2000, 66: 1963-8.
22. Heied CA, Stevens J, Livak KJ, Williams PM: Real time PCR. Genome Res. 1996, 6: 986-94.
23. Matsubara Y, et al.: A fluorogenic allele-specific amplification method for DNA-based screening for inherited metabolic disorders. Acta Paediatr Suppl. 1999, 88: 65-8.
otrzymano/received: 2008-04-23 zaakceptowano/accepted: 2008-05-15 Adres/address: *Grzegorz Kurzawski Międzynarodowe Centrum Nowotworów Dziedzicznych. Zakład Genetyki i Patomorfologii, Pomorska Akademia Medyczna ul. Połabska 4, 70-115 Szczecin tel.: (0-91) 466-15-32 e-mail: gkurz@sci.pam.szczecin.pl The whole paper Analizy molekularne DNA i RNA w wykrywaniu dziedzicznych predyspozycji do nowotworów is also available at On-line Medical Library. |
  Please click on selected cover
 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||






Chcesz być na bieżąco? Polub nas na Facebooku: strona Wydawnictwa na Facebooku |