|
© Borgis - Postępy Nauk Medycznych 7, s. 427-430
*Tadeusz Dębniak, Jan Lubiński
Zasady dziedziczenia predyspozycji do nowotworów
Principles of genetic predisposition to malignancies
Międzynarodowe Centrum Nowotworów Dziedzicznych Zakład Genetyki i Patomorfologii Pomorska Akademia Medyczna, Szczecin
Kierownik Zakładu Genetyki i Patomorfologii: prof. dr hab. med. Jan Lubiński Streszczenie
Nowotwory złośliwe powstają w wyniku genetycznie uwarunkowanej predyspozycji oraz, w różnym stopniu, wpływu czynników środowiskowych. Czynniki genetyczne są odpowiedzialne za predyspozycję jednogenową oraz wielogenową, „wysoką” oraz „umiarkowaną”. Szacuje się, że około 30% wszystkich nowotworów powstaje w wyniku wysokiej, genetycznie uwarunkowanej predyspozycji. W zależności od rodzaju uszkodzeń i miejsca uszkodzenia DNA różny jest typ dziedziczenia oraz charakterystyka rodowodowo-kliniczna. Rodowodowo dziedziczenie jednogenowe autosomalne dominujące na ogół charakteryzuje występowanie zachorowań w każdym kolejnym pokoleniu (pionowa transmisja), zarówno u mężczyzn jak i u kobiet; u blisko 50% krewnych. W tym typie chorób genetycznych mutacje konstytucyjne (tj. obecne we wszystkich komórkach organizmu) w pojedynczym genie są główną przyczyną zachorowania. Oceniając rodowód rodziny obciążonej nowotworami należy również pamiętać o tzw. fenokopiach – pojawianiu się przypadkowych, niezwiązanych z nosicielstwem danej mutacji zachorowań na nowotwory w rodzinie. W przypadku wielogenowej predyspozycji na nowotwory zachorowuje zwykle tylko pojedyncza osoba w rodzinie. Wielogenowa predyspozycja do nowotworów może być związana z wysokim lub umiarkowanie zwiększonym ryzykiem zachorowania. Wydaje się, że uszkodzenia DNA umiarkowanie zmieniające ryzyko zachorowania odpowiadają głównie za mało nasilone rodzinne agregacje zachorowań. Taki patomechanizm może mieć ważne znaczenie kliniczne, bowiem słaba rodzinna agregacja nowotworów jest częsta. Współdziałanie „słabych” mutacji i polimorfizmów w wielu genach oraz dodatkowo wpływ czynników środowiskowych może znacząco zwiększać ryzyko nowotworów złośliwych różnych narządów. Słowa kluczowe: dziedziczenia jednogenowe, dziedziczenia wielogenowe
Summary
Human malignancies are caused by genetic factors and gene-environment interactions. Genetic susceptibility can be divided into monogenic and polygenic predisposition, the risk of cancer development can be defined as "high” or "moderate”. It is estimated that around 30% of all malignancies are caused by "high-risk” genetic predisposition. Depending on the position and type of DNA changes the pattern of inheritance as well as clinical and pedigree characteristics may be different. Pedigrees of monogenic diseases with autosomal dominant inheritance are characterised by occurrence of disorder in all generations (vertical transmission), among men and women, among 50% of the relatives. Such diseases are caused by constitutional mutations of single genes, present in all cells of the body. Evaluation of the pedigree and clinical data of families with aggregations of cancers should exclude phenocopies (accidental malignancy not related to mutation responsible for the aggregation of malignant tumors). In polygenic type of inheritance single individuals can be affected. DNA alterations associated with "moderate” risk are responsible for moderate cancer familial aggregations. Such patomechanism may have significant clinical impact, since moderate cancer familial aggregations are frequent. Associations of "weak” mutations and polymorphisms of many genes and additional influence of environmental factors can significantly increase the risk of cancer development. Key words: monogenic predisposition, polygenic predisposition
Szacuje się, że około 30% wszystkich nowotworów powstaje w wyniku wysokiej, genetycznie uwarunkowanej predyspozycji (1). Świadczą o tym przede wszystkim analizy zgodności zachorowań wśród bliźniaków jednojajowych, a więc identycznych genetycznie. Jeśli jeden z nich zachoruje np. na raka prostaty czy piersi, to prawdopodobieństwo, że drugi bliźniak będzie dotknięty nowotworem tego samego narządu wynosi odpowiednio ok. 40 i 30% (2). Ponadto, jeśli uwzględnimy również przypadki zachorowań na nowotwory, ale o innej lokalizacji, np. raka piersi u jednej osoby, a raka żołądka u drugiej, to zgodność wśród bliźniaków jednojajowych jest jeszcze wyższa.
Przyjmuje się, że nowotwory powstają najczęściej w wyniku genetycznej predyspozycji jednogenowej lub wielogenowej.
Jednogenowa predyspozycja do nowotworów
W tym typie chorób genetycznych mutacje konstytucyjne (tj. obecne we wszystkich komórkach organizmu) w pojedynczym genie są główną przyczyną zachorowania. We wszystkich nowotworach występują mutacje genów. Są to jednak w zdecydowanej większości tak zwane mutacje somatyczne, które od konstytucyjnych różnią się tym, że występują jedynie w tkance nowotworowej. Na obecnym etapie rozwoju nauki, wśród dziedzicznych przyczyn nowotworów diagnozowanych w celach praktycznego usprawnienia opieki medycznej niemal wyłącznie rozpoznawana jest predyspozycja jednogenowa z rodowodowymi cechami dziedziczenia autosomalnie dominującego. Według modelu dziedziczenia Mendla każdy osobnik ma dwie kopie (allele) genu odpowiedzialnego za daną cechę lub chorobę, zajmujące identyczną pozycję (lokus) na homologicznych chromosomach. Każde dziecko otrzymuje jedną kopię genu od ojca, drugą zaś od matki. Zgodnie z zasadami dziedziczenia jednogenowego dominującego, posiadanie zaburzenia – wrodzonej mutacji już tylko w jednej kopii genu powoduje predyspozycję do zachorowania (3). Reguła ta jest zachowana wówczas, gdy gen związany z rozwojem nowotworów dziedzicznych jest protoonkogenem, jak np. RET, którego mutacje konstytucyjne predysponują do zespołu MEN2. W większości nowotwory powstają u nosicieli mutacji w tzw. genach supresorowych (antyonkogenach) jak np. p53, czy w genach naprawy uszkodzeń DNA jak np. MSH2, które molekularnie mają charakter recesywny. U nosicieli mutacji takich genów dochodzi do rozwoju nowotworów, dlatego, że z bardzo wysoką częstością u tych osób występuje w ciągu życia inaktywacja drugiego allela (najczęściej poprzez delecję) (4).
Rodowodowo dziedziczenie jednogenowe autosomalne dominujące charakteryzuje występowanie zachorowań:
a) w każdym kolejnym pokoleniu (pionowa transmisja);
b) zarówno u mężczyzn jak i u kobiet;
c) u blisko 50% krewnych (5).
Przykładowy rodowód przedstawiono na rycinie 1. W rodzinie tej obserwuje się też coraz młodszy wiek pojawiania się nowotworów w każdym kolejnym pokoleniu (tak zwana antycypacja).
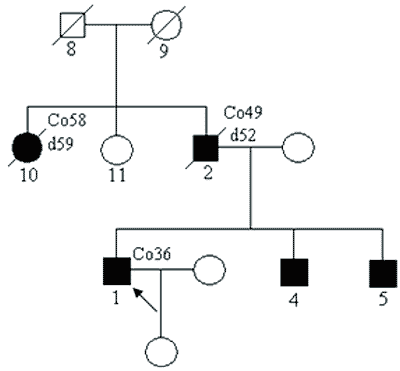 Ryc. 1. Rodzina z zespołem Lyncha – z rodowodowymi cechami choroby jednogenowej autosomalnie dominującej.
Cech charakterystycznych dla dziedziczenia jednogenowego dominującego nie stwierdza się w sytuacjach:
a) mutacji germinalnych (tj. w plemnikach lub komórkach jajowych) „de novo” – w takich przypadkach nie występują zachorowania w pokoleniach przodków probanta (osoby zasięgającej porady genetycznej); mutacja jest przekazywana na następne pokolenia – rycina 2;
 Ryc. 2. Rodowód rodziny z chorobą wywołaną mutacją germinalną de novo w obrębie genu VHL.
b) mutacji mozaikowych, tj. obecnych tylko w niektórych tkankach; mutacje takie powstają zwykle „de novo” w okresie zarodkowym; w takich przypadkach obserwuje się zachorowania u pojedynczej osoby w rodzinie, a mutacja jest przekazywana na następne pokolenia tylko wówczas, gdy występuje w komórkach germinalnych;
c) mutacji o niskiej penetracji; penetrację definiujemy jako stosunek liczby osób chorych do liczby nosicieli mutacji; w zespołach predyspozycji do nowotworów sięga ona nawet 0,8-0,9; w przypadkach mutacji o niskiej penetracji jest on dużo niższy i wówczas w rodzinie chorują tylko pojedyncze osoby (6); przykładowy rodowód pokazano na rycinie 3;
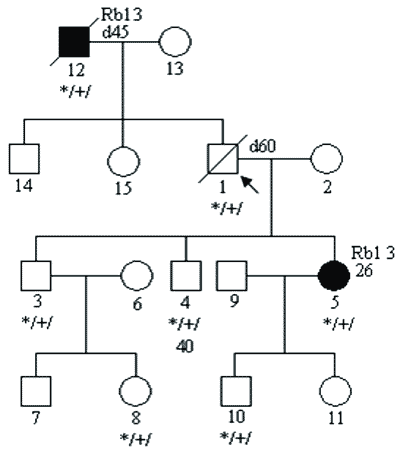 Ryc. 3. Rodowód rodziny z niską penetracją mutacji genu Rb1.
d) mutacji skutkujących zachorowaniem tylko u jednej płci; na przykład nosicielami mutacji BRCA1są zarówno mężczyźni jak i kobiety, na raka jajnika chorują jednak tylko kobiety z tą zmianą;
e) w „małych” rodzinach z niewielką liczbą osób.
Oceniając rodowód rodziny obciążonej nowotworami należy również pamiętać o tzw. fenokopiach – pojawianiu się przypadkowych, niezwiązanych z nosicielstwem danej mutacji zachorowań na nowotwory w rodzinie (7). Na poniższej rycinie przedstawiono przykładowy rodowód – z 4 rakami piersi wywołanymi dziedziczną mutacją genu BRCA1oraz 1 rakiem piersi u osoby niebędącej nosicielem tej mutacji. U potomstwa tej pacjentki nie stwierdza się wysokiego ryzyka związanego z mutacją BRCA1.
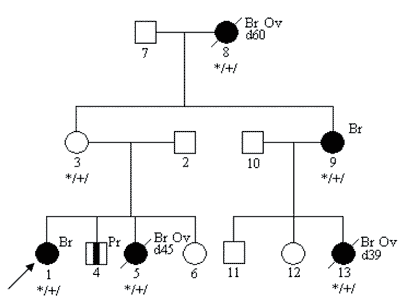 Ryc. 4. Fenokopia – rak piersi niezwiązany z mutacją konstytucyjną w rodzinie z mutacją BRCA1.
Legenda do rodowodów przedstawionych na rycinach 1-4: Co36 – rak jelita grubego rozpoznany w 36 roku życia d52 – zgon w 52 roku życia Rb1 3 – siatkówczak rozpoznany w 3 roku życia Hb – „hemangioblastoma” siatkówki Ov – rak jajnika Pr – rak prostaty */+/ – nosiciel mutacji konstytucyjnej */-/ – osobnik niebędący nosicielem mutacji konstytucyjnej Wielogenowa predyspozycja do nowotworów
W przypadku tego typu predyspozycji na nowotwory zachorowuje zwykle tylko pojedyncza osoba w rodzinie. Wielogenowa predyspozycja do nowotworów może być związana z wysokim lub umiarkowanie zwiększonym ryzykiem zachorowania.
W wyniku prowadzonych w naszym Ośrodku badań jak dotąd poznano geny/mutacje i polimorfizmy zwiekszające (na ogół umiarkowanie) ryzyko nowotworów w ponad 90% nieselekcjonowanych raków piersi (96% raków piersi rozpoznanych po 50. roku życia, 99% raków zrazikowych – tabela 1, 2, 3), 89% raków jelita grubego, 72% czerniaków złośliwych, 36% raków jajnika oraz 27,5% raków prostaty. Wydaje się, że uszkodzenia DNA umiarkowanie zmieniające ryzyko zachorowania odpowiadają głównie za mało nasilone rodzinne agregacje zachorowań. Taki patomechanizm może mieć ważne znaczenie kliniczne, bowiem słaba rodzinna agregacja nowotworów jest częsta.
Tabela 1. Częstość markerów zidentyfikowanego panelu w nieselekcjonowanych kolejnych rakach piersi i kontrolach (9).
Tabela 2. Częstość markerów zidentyfikowanego panelu w rakach zrazikowych i kontrolach (9).
Tabela 3. Częstość markerów zidentyfikowanego panelu w rakach rozpoznanych w wieku powyżej 50 r.ż. i kontrolach (9).
Współdziałanie „słabych” mutacji i polimorfizmów w wielu genach oraz dodatkowo wpływ czynników środowiskowych może znacząco zwiększać ryzyko nowotworów złośliwych różnych narządów. Współdziałanie linearne (akumulacja zmian) występuje, jeśli całkowite ryzyko zachorowania na nowotwór składa się z sumy ryzyk związanych z poszczególnymi „słabymi” mutacjami w wielu genach, na przykład w przypadku dwukrotnego wzrostu ryzyka zachorowania u nosiciela mutacji w genie A oraz mutacji w genie B (jeśli obie te mutacje są związane z 50% wzrostem ryzykiem zachorowania). W innych przypadkach występuje współdziałanie nieliniowe (interakcja zmian) – na przykład przy pięciokrotnym wzroście ryzyka u nosiciela wyżej wymienionych mutacji (8). Piśmiennictwo
1. Lynch HT, Fusaro RM, Lynch J: Hereditary cancer in adults. Cancer Detect Prev 1995, 19: 219-33.
2. Lichtenstein P, et al.: The Swedish Twin Registry: a unique resource for clinical, epidemiological and genetic studies. J Intern Med 2002, 252: 184-205.
3. Knudson A: Mutation and cancer: a statistical study. Prc Natl Acad Sci USA 1971, 68: 820-3.
4. Friedman JM: Genetics and epidemiology, congenital anomalies and cancer. Am J Hum Genet 1997, 60: 469-73.
5. Marra G, Boland CR: Hereditary nonpolyposis colorectal cancer-the syndrome, the genes, historical perspectives. J Natl Cancer Inst 1995, 87: 1114-24.
6. Lubiński J: Hereditary tumors – prophylactics, early diagnosis, treatment. Biotechnologia 1996, 35: 202-7.
7. Friedman JM, et al.: Genetyka, wydanie I polskie pod redakcją J. Limona. Elsevier Urban & Partner, Wrocław 2000.
8. Cybulski C, et al.: Epistatic relationship between the cancer susceptibility genes CHEK2 and p27. Cancer Epidemiol Biomarkers Prev. 2007, 16: 572-6.
9. Lubinski J, et al.: Genetic contribution to all cancers: the first demonstration using the model of breast cancers from Poland stratified by age at diagnosis and tumour pathology, Breast Cancer Res Treat 2008.
otrzymano/received: 2008-04-23 zaakceptowano/accepted: 2008-05-15 Adres/address: *Tadeusz Dębniak Centrum Nowotworów Dziedzicznych. Zakład Genetyki i Patomorfologii, Pomorska Akademia Medyczna ul. Połabska 4, 70-115 Szczecin tel.: (0-91) 466-15-32 e-mail: debniak@wp.pl The whole paper Zasady dziedziczenia predyspozycji do nowotworów is also available at On-line Medical Library. |
  Please click on selected cover
 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||






Chcesz być na bieżąco? Polub nas na Facebooku: strona Wydawnictwa na Facebooku |