|
© Borgis - Postępy Nauk Medycznych 6, s. 413-419
*Dariusz Chmielewski
Choroba Pageta kości
Paget´s disease of bone
Zespół Ortopedyczno-Traumatologiczny Szpitala HOSPITEN – Lanzarote, Puerto del Carmen/Hiszpania
Kierownik: dr n. med. Dariusz Chmielewski Streszczenie
Choroba Pageta kości należy do rzadziej występujących chorób metabolicznych tkanki kostnej. Jej przyczyną jest zaburzenie funkcjonowania osteoklastów. Choroba rozwija się fazowo, a objawy kliniczne i radiologiczne zmieniają się w czasie trwania schorzenia. Ich właściwa interpretacja oraz stwierdzenie określonych zaburzeń w zakresie badań biochemicznych jest podstawą do postawienia rozpoznania. Wiele innych patologii powoduje zaburzenia podobne do choroby Pageta kości, sprawiając, że pomyłki diagnostyczne nie są rzadkością. Dzięki wprowadzeniu do leczenia nowych leków (głównie bisfosfonianów) możliwe jest lepsze kontrolowanie zaburzeń metabolizmu kostnego występującego w czasie trwania schorzenia oraz uzyskanie dłuższych okresów remisji. Innym celem leczenia tej choroby jest zmniejszenie dolegliwości bólowych oraz jej następstw, czyli głównie zniekształceń kości i zwyrodnień stawów. Coraz szerzej stosowane u pacjentów z chorobą Pageta kości leczenie operacyjne przynosi dobre wyniki, jeśli połączone jest z leczeniem farmakologicznym, przy zachowaniu stałej kontroli metabolicznej. Słowa kluczowe: choroba Pageta kości, osteitis deformans, epidemiologia, objawy, diagnostyka, leczenie
Summary
Paget´s disease of bone is a rare metabolic bone disease. It is caused by disturbances in osteoclasts´ function. The disease is characterized by phase evolution with alternating radiological and clinical symptoms. Its correct interpretation and confirmation with particular variations in biochemical markers is crucial for diagnosis. Many other pathologies may mimic Paget´s disease of bone. Diagnostic mistakes are not rare. Thanks to wide disposal of new drugs (especially bisphosphonates) better control of disturbances of bone metabolism and longer remission periods are possible. The other aim of the treatment is diminishing of pain and prevention of the disease´s complications – joint degradation and bone deformations. Widely used operative treatment is beneficial if connected with pharmacological intervention and continuous biochemical control Key words: Paget´s disease of bone, osteitis deformans, epidemiology, symptoms, diagnostics, treatment
Choroba Pageta kości (PD) jest ograniczoną, ogniskową chorobą metaboliczną szkieletu, charakteryzującą się nadaktywną przebudową kostną z przewagą procesów resorpcyjnych, okresowo z wyraźnie zwiększoną osteogenezą, doprowadzającą do znacznego zaburzenia struktury i kształtu kości.
Została ona opisana po raz pierwszy w 1876 roku przez sir Jamesa Pageta (1814-1889). W „Transactions” – periodyku Królewskiej Szkoły Medycyny, podał on rozważania i wnioski dotyczące, jak sam podaje, rzadkiej choroby kości. Nazwał ją „ osteitis deformans ”, czyli zniekształcające zapalenie kości, ponieważ był przekonany, że istotą problemu jest proces zapalny toczący się w tkance kostnej, wywoływany przez nieznany czynnik. Dzisiaj wiadomo, że te założenia są nieprawidłowe, ale wiele z Jego spostrzeżeń okazało się niezwykle trafnych i nawet dzisiaj stanowią podstawę opisu zmian w przebiegu tej jednostki chorobowej. Nawet nazwa „osteitis deformans” nadal często pojawia się w piśmiennictwie.
Choroba Pageta kości występuje szczególnie często w krajach Europy Północnej [1]. Badania radiologiczne przeprowadzone w Wielkiej Brytanii w latach 70. wykazały, że występowanie choroby może sięgać nawet 5,4% w populacji osób powyżej 55 roku życia [2, 3]. Stwierdzono jednocześnie wyraźną korelację częstości występowania choroby i wieku. I tak, w grupie po 85 roku życia częstość występowania choroby jest prawie 5-krotnie wyższa w porównaniu z grupą osób poniżej 60 roku życia [4]. Podobna analiza przeprowadzona w Stanach Zjednoczonych wykazała niższą częstość występowania choroby, bo 2,3% w populacji między 65 a 74 rokiem życia [5]. Ocenia się, że na terenie Anglii i Walii chorych jest 118 000 kobiet i 144 000 mężczyzn [6]. Tym samym potwierdzają się dane o proporcji w rozkładzie zachorowań w obu płciach – częściej chorują mężczyźni, a stosunek mężczyźni/kobiety wynosi 1,5/1. Udowodniono również zwiększone rodzinne ryzyko zachorowania – jest ono 3-7 razy większe niż w całej populacji [7].
W Polsce brak jest danych na temat rozpowszechnienia choroby.
Patrząc na rozkład częstości występowania PD w skali całego świata, zwraca uwagę jej bezpośredni związek z krajami historycznie powiązanymi z Anglią, czyli na wschodnim wybrzeżu Stanów Zjednoczonych, w Kanadzie, Australii i Nowej Zelandii [8].
Patogeneza choroby wiązana jest z nadmierną aktywacją osteoklastów (OC). Sugerowano, że proces ten jest wynikiem przebytego zakażenia wirusowego.
W 1977 Rebel opublikował wyniki własnych prac, w których wykrył nietypowe wtręty w obrębie jąder i cytoplazmy osteoklastów pochodzących od pacjentów z PD [9]. Stwierdzono, że były one podobne do nukleokapsydów paramyksowirusów. Znajdowano jednak również materiał typowy dla innych rodzin wirusów, takich jak wirus odry (MV) lub RSV (respiratory syncytial virus) [10]. Dwa lata później podobne informacje podał kontynuując te badania Basle, donosząc o stwierdzeniu cząstek wirusa odry i innych paramyksowirusów [11]. Współpracujący z Rebelem Mills potwierdził w 1984 roku obecność antygenów wirusa odry i RSV (respiratory syncytial wirus) w osteoklastach pacjentów z osteitis deformans i ich brak w przypadku innych chorób metabolicznych kości, wykorzystując techniki immunocytochemiczne [12]. Pojawiały się też inne próby wyjaśnienia procesu powstawania charakterystycznych dla choroby Pageta, zmian kostnych. Cytowany przez Resnicka i Niwayamę [13] Henneman opublikował w 1964 roku pracę, w której dowodził, że istotą schorzenia jest uogólniona, wrodzona patologia tkanki łącznej. Idea ta jednak szybko upadła i większość znawców tematu powróciła do teorii pierwotnej infekcji wirusowej [14].
Badania przeprowadzone w ostatnich latach istotnie poszerzyły wiedzę na temat etiopatogenezy choroby Pageta kości. Wykazano, że osoby z PD, szczególnie w jej postaci wieloogniskowej, charakteryzują się znacznie podwyższonym stężeniem RANKL (Receptor Activator of Nuclear Factor-kB Ligand) oraz, w mniejszym stopniu, osteoprotegeryny (OPG) w surowicy krwi [15]. Wykazano również podwyższone stężenie interleukiny-6 w surowicy krwi w tej grupie pacjentów [16]. Stwierdzono ponadto, że u chorych z PD komórki zrębu kości wykazują obecność RANKL i stymulują aktywność OC za pośrednictwem receptora RANK (Receptor Activator of Nuclear Factor-kB) [17].
Z drugiej strony osteoklasty osób chorych na PD wykazują nadmierną reakcję na RANKL oraz na aktywny metabolit witaminy D: 1,25 (OH)2 D3 [18,19]. Komórki te charakteryzują się także zwiększoną ekspresją TAFII-17, składnika kompleksu transkrypcyjnego TFIID, który pełni rolę koaktywatora transkrypcji mediowanej przez receptor witaminy D [20].
Istotną rolę w patogenezie PD przypisuje się czynnikom genetycznym. Opisano liczne rodziny, w których choroba dziedziczyła się w sposób autosomalny dominujący [21, 22].
Zidentyfikowano co najmniej 4 mutacje genowe predysponujące do rozwoju PD. Wykazano, że rozwijająca się w młodym wieku rodzinna postać choroby związana jest z mutacją w TNFRSR11A kodującym RANK, zaś polimorfizm genu TNFRSR11B, który koduje OPG, zwiększa ryzyko wystąpienia klasycznej postaci PD. Najistotniejsze znaczenie kliniczne mogą mieć jednak mutacje genu p62 (sekwestozomu 1). Ich obecność stwierdzono u około 30% pacjentów z rodzinną chorobą Pageta, a także u części chorych ze sporadyczną postacią PD [22, 23]. Stwierdzono, że p62 odgrywa wiodącą rolę w aktywacji RANK przez m.in. TNF-α i IL-1, za pośrednictwem interakcji z atypowymi kinazami białkowymi ζPKC i λPKC [24,25]. Wykazano, że u pacjentów z mutacją w genie p62 dochodzi do namnażania się preosteoklastów przy niższych stężeniach RANKL, TNF- α i 1,25 (OH)2 witaminy D3 niż u osób zdrowych [26].
Rola tej mutacji pozostaje jednak niejasna, gdyż nie u wszystkich osób ze zmianami w obrębie p62 dochodzi do rozwoju PD [27, 28, 29]. Sugeruje się, że czynniki genetyczne, takie jak mutacja p62, mogą nasilać proces tworzenia się osteoklastów, nie powodują jednak powstania patologicznych OC i charakterystycznych ognisk patologicznej przebudowy kości [26].
Mimo lepszego zrozumienia etiopatogenezy PD, choroba stanowi nadal ogromny problem kliniczny i wciąż pozostaje nieuleczalna. W blisko 70% przypadków przebiega bezobjawowo, co utrudnia postawienie właściwego rozpoznania [30]. Chociaż dysponujemy wieloma środkami dającymi możliwość osiągnięcia i utrzymania długotrwałej remisji, istnieją trudności w ustaleniu, który z pacjentów powinien zostać poddany terapii. Biorąc pod uwagę liczbę pacjentów z chorobą Pageta kości, którzy są zagrożeni wystąpieniem powikłań, i fakt, że wielu z nich wymagać będzie wielokrotnych kursów leczenia i wieloletniego monitorowania, nietrudno zauważyć problem finansowy z tym związany. Nie jest on ograniczony tylko do krajów z wysokim wskaźnikiem zachorowalności dla tej choroby. Jak podkreśla amerykańska Fundacja Pageta [2], problem nabrał wymiaru międzynarodowego, a może nawet globalnego. Rosnąca migracja z jednej strony i koszty opieki nad pacjentami z drugiej strony nie mogą mieć charakteru lokalnego. Konieczne są poszukiwania rozwiązań systemowych, a przede wszystkim unifikowanie postępowania w postaci jasnych i precyzyjnych zbiorów zasad postępowania diagnostycznego i terapeutycznego.
Aktualne wskazania do leczenia opierają się na teoretycznych podstawach i ustalane są empirycznie. Nadal istnieją rozbieżności, nawet wśród wybitnych znawców zagadnienia, dotyczące postępowania w konkretnych przypadkach klinicznych. Problemem jest wybór leku, ustalenie jego dawki, schemat podawania i kontroli efektów terapii oraz postępu choroby. Dlatego celowe wydaje się przeprowadzenie gruntownej, wielokierunkowej analizy materiału w ośrodkach zajmujących się tym tematem oraz prezentacja i popularyzacja wyników tych opracowań oraz wynikających z nich wniosków [31].
Złożonym problemem klinicznym jest transformacja nowotworowa w kierunku mięsaka kostnopochodnego w PD. Według niektórych autorów ryzyko zezłośliwienia ogniska jest kilka tysięcy razy większe niż wystąpienia pierwotnego mięsaka kości w populacji ogólnej [31].
W obrazie klinicznym choroby dominują bóle (kostne i stawowe), deformacje (łukowate wygięcia kości długich oraz deformacje czaszki), złamania patologiczne („pełne” złamania z przemieszczeniem odłamów i nieprzemieszczone linijne pęknięcia o typie „złamań beleczki kredy”), objawy neurologiczne (głuchota, objawy uciskowe nerwów twarzowych, zespoły ciasnoty kanału kręgowego, wtrętowe zapalenie mięśni kończyn dolnych) oraz transformacja nowotworowa [32, 33].
PD rozwija się powoli, a w wielu przypadkach, przez wiele lat nie wykazuje żadnych klinicznych cech progresji. Wystąpienie objawów, głównie dolegliwości bólowych i znacznych deformacji z wtórnymi zmianami zwyrodnieniowymi w stawach tworzonych przez zajęte kości, jest uciążliwe dla pacjenta i najczęściej jest momentem podjęcia diagnostyki i postawienia właściwego rozpoznania klinicznego [33].
Choroba Pageta może obejmować jedną kość (lokalizacja jednoogniskowa) lub kilka miejsc szkieletu (lokalizacja wieloogniskowa). Kości zajęte są pierwotnie i nie obserwuje się rozprzestrzeniania procesu na inne lokalizacje w trakcie rozwoju choroby [32]. Najczęstszą lokalizacją zmian są kości miednicy i kość krzyżowa (ponad 60% przypadków), następnymi w kolejności – kręgosłup (50%), czaszka i kość udowa (40%), kość piszczelowa, ramienna i obojczyk (20%). Kości rąk i stóp oraz twarzoczaszka są rzadko zajęte [32].
Rozważania na temat diagnostyki choroby Pageta kości należy rozpocząć od omówienia możliwości obrazowania w tym zakresie. Od czasów sir Jamesa Pageta bowiem charakterystyczny obraz kości zajętych tym procesem jest podstawą do postawienia rozpoznania [13]. Jest on na tyle typowy, że Borejko i Dziak [32] w rozdziale na temat niektórych rzadkich chorób kości piszą: „Obraz radiologiczny jest zwykle charakterystyczny i rozpoznanie ustala się zwykle na pierwszy rzut oka”. Istnieją jednak przypadki, w których obraz ten nie jest tak typowy i konieczne jest przeprowadzenie wnikliwego różnicowania, często z wykorzystaniem innych technik obrazowania, jak tomografia komputerowa lub tomografia jądrowego rezonansu magnetycznego.
Objawy radiologiczne występują fazowo i tym samym wiążą się z określeniem etapu rozwoju choroby. W pierwszym okresie choroby dochodzi do aktywacji osteoklastów, co powoduje miejscową stymulację procesów resorpcyjnych w tkance kostnej. W praktyce klinicznej ten okres rozwoju choroby określa się jako fazę resorpcyjną lub rzadziej – fazę lityczną [13]. W obrazie radiologicznym stwierdzamy wówczas typowe ogniska osteolityczne z zanikiem utkania beleczkowego kości [13, 32]. Początkowo mają one wyraźnie ograniczony charakter (korowe ubytki „przecinkowate”), umiejscawiają się głównie w okolicach końców stawowych kości długich i stopniowo rozprzestrzeniają się w kierunku trzonów. Po pewnym czasie ulegają połączeniu w większe twory i wówczas układają się w postaci ubytków korowych o kształcie litery V („objaw ociekającego wosku”).
Na tym samym etapie rozwoju choroby w przypadku zajęcia czaszki stwierdzić można zmiany zanikowe opisywane jako osteoporosis circumscripta - plackowate ogniska ubytkowe, zlokalizowane głównie z zakresie blaszki zewnętrznej kości potylicznej lub rzadziej kości czołowej (ryc. 1) [34].
 Ryc. 1. Osteoporosis circumscripta – wczesna faza choroby Pageta kości, zwraca uwagę charakterystyczny plackowaty ubytek w obrębie blaszki zewnętrznej pokrywy czaszki, stwierdzany w obrazie rentgenowskim jako przejaśnienie cienia kostnego w obrębie kości czołowej i częściowo skroniowej (materiał własny, +RM, lat 49).
W kolejnym etapie, nadal w fazie resorpcyjnej, proces niszczenia tkanki kostnej doprowadza do powstania zlewnych ognisk ubytkowych. Pojawiają się typowe zmiany radioprzezierne o typie torbielowatych ubytków. Mogą być one miejscem złamań patologicznych z powodu osłabienia wytrzymałości tkanki kostnej [34].
Odczynowe zwiększenie aktywności kościotwórczej charakteryzuje następny okres rozwoju choroby, określany jako faza mieszana. Niekontrolowany proces osteogenezy powoduje pogrubienie beleczek kostnych, zaburzenie ich przebiegu i rozmieszczenia oraz postępujące pogrubienie warstwy korowej. Dochodzi do stopniowego poszerzania się zewnętrznych wymiarów kości, a niesymetryczny charakter tego procesu powoduje powstawania zaburzeń osi i deformacji. W obrębie miednicy zmiany te określa się często jako wełnisty rysunek beleczkowy (ryc. 2). Najczęściej jednak używanym określeniem jest porównanie do kłębków waty lub bawełny (ang. cotton wool) (ryc. 3).
 Ryc. 2. Wełnisty obraz utkania kostnego w obrębie kości kulszowej miednicy – faza mieszana choroby Pageta kości (materiał własny, + KP, lat 71).
 Ryc. 3. Obraz typu „kłębki waty” – faza mieszana choroby Pageta kości (materiał własny, +WH, lat 72).
Niekiedy w obrębie czaszki można napotkać obraz naprzemiennie występujących drobnych ognisk resorpcyjnych i osteosklerotycznych, dający charakterystyczne „pstre” utkanie sklepienia. Zjawisko to określa się jako „obraz pieprzu i soli”.
Innym, rzadkim objawem radiologicznym jest hiperostoza kości sklepienia czaszki. Występuje ona wówczas, gdy tworzenie kości nie było poprzedzone obecnością zmian resorpcyjnych i charakteryzuje się wyraźnym pogrubieniem oraz sklerotyzacją całej lub części pokrywy czaszki (ryc. 4).
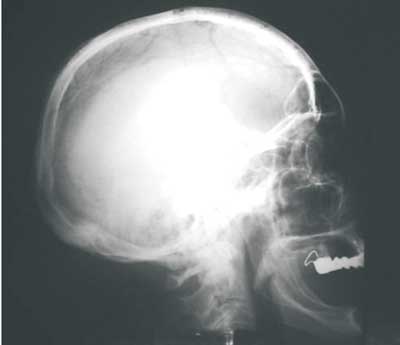 Ryc. 4. Hiperostoza czaszkowa – faza sklerotyczna choroba Pageta kości; zwraca uwagę znaczne pogrubienie pokrywy czaszki w obrębie kości czołowej (materiał własny, + MS, lat 55).
W późnym okresie fazy mieszanej, w przypadku zajęcia kręgosłupa, stwierdza się najbardziej typowy obraz radiologiczny PD – krąg ramowy (ang. picture frame vetrebra) (ryc. 5). Zgrubienie (zdwojenie) blaszek granicznych daje obraz grubej, podwójnej ramki otaczającej wnętrze trzonu kręgowego. Charakterystyczne jest, że zajęty przez chorobę Pageta krąg nie ulega rozdęciu i nie zwiększa swoich wymiarów zewnętrznych. Tkanki otaczające go, przede wszystkim sąsiadujące z nim tarcze międzykręgowe, ulegają stopniowemu przerostowi na podłożu zmian zapalnych. Ten właśnie proces jest odpowiedzialny za powstawanie zespołów uciskowych ze stenozą kanału kręgowego i otworów międzykręgowych włącznie [34].
 Ryc. 5 – krąg ramowy – faza mieszana choroby Pageta kości
(materiał własny, + ST, lat 63) Innym zjawiskiem radiologicznym stwierdzanym w obrębie kręgosłupa jest nadmierna sklerotyzacja kręgów, dająca obraz kręgu „z kości słoniowej” (ang. ivory-like vertebra) (ryc. 6) [34].
 Ryc. 6. Obraz „kręgu z kości słoniowej” – faza sklerotyczna choroby Pageta kości (materiał własny, + KP, lat 71).
Stwierdzenie tego objawu pozwala na rozpoznanie trzeciej (końcowej) fazy choroby – fazy sklerotycznej. Objawy pogrubienia warstwy korowej występują także w kościach długich, dając razem z nieregularnym układem beleczek kostnych typowy dla choroby Pageta obraz radiologiczny.
Jak wynika z wytycznych zawartych w Zaleceniach postępowania w chorobie Pageta kości, opublikowanych przez brytyjską grupę roboczą pod kierunkiem Selby´ego, Davie, Ralstona i Stone´a [33] każdorazowo rozpoznanie choroby powinno być potwierdzone radiologicznie, przynajmniej w jednej lokalizacji w szkielecie. Dodatkowo zaleca się wykonywanie zdjęć rentgenowskich wszystkich miejsc bolesnych, natomiast radiogramy seryjne kośćca nie powinny być wykonywane.
W przebiegu choroby najistotniejszą rolę pełni nadmierna aktywacja procesów resorpcji i tworzenia tkanki kostnej. Ocena aktywności specyficznych markerów biochemicznych przemian kostnych, innych niż fosfataza alkaliczna, ma duże znaczenie w pierwotnej diagnostyce [33]. Odnosi się to szczególnie do wczesnej, bezobjawowej fazy choroby, kiedy indukowana jest osteoklastyczna resorpcja. Według brytyjskiego Narodowego Stowarzyszenia na rzecz Złagodzenia Choroby Pageta (National Association for the Relief of Paget´s Disease) obniżenie aktywności fosfatazy alkalicznej o 25% uznawane jest za znaczącą odpowiedź na leczenie farmakologiczne [33]. Według Tiegsa 20-25% wzrost ponad wcześniej oznaczoną wartość fosfatazy alkalicznej powinien stanowić wskazanie do wdrożenia terapii antyresorpcyjnej ze spodziewanym efektem, mierzonym obniżeniem aktywności fosfatazy alkalicznej, na tym samym poziomie [35]. Zakres od 75-80% do 120-125% od oznaczonej wartości fosfatazy alkalicznej Tiegs określa jako wartości progowe.
W analizie aktywności procesu chorobowego, a w szczególności w ustalaniu wskazań do leczenia, monitorowaniu skuteczności terapii oraz ocenie ewentualnej progresji choroby pomocne są inne niż fosfataza alkaliczna markery obrotu kostnego [30, 33, 34].
Spośród dostępnych markerów obrotu kostnego w praktyce klinicznej wykorzystuje się oznaczenia jednego z nich w celu określenia tempa tworzenia i jednego dla oceny resorpcji kości.
W monograficznym opracowaniu na temat strategii leczenia choroby Pageta kości, Meunier i Vignot już w 1995 roku [34] podkreślili konieczność rozdzielenia dwóch rodzajów terapii:
– krótkoterminowej – jej celem jest szybkie zmniejszenie dolegliwości bólowych lub objawów neurologicznych występujących w przebiegu choroby – oraz
– długoterminowej – ocenianej zmniejszeniem aktywności metabolicznej tkanki kostnej; jej celem jest zmniejszenie ryzyka wystąpienia deformacji, złamań i wtórnych zmian zwyrodnienionych, a także bólu i nowotworzenia.
Cele kompleksowego leczenia choroby Pageta kości określa w swoich wytycznych Selby wraz z zespołem [33]. Zalicza do nich:
– ograniczenie bólów kostnych związanych z ogniskiem patologicznym; inne dolegliwości bólowe są wskazaniem do leczenia objawowego,
– zmniejszenie ryzyka złamania na tle zmian chorobowych; wpływ leczenia antyresorpcyjnego włączonego po wystąpieniu złamania na przyspieszenie gojenia nie został udowodniony,
– zapobieganie deformacjom układu kostnego, wpływ leczenia jest tu niejasny; wykazano możliwość zapobiegania deformacjom twarzoczaszki dzięki leczeniu z wykorzystaniem bisfosfonianów,
– zapobieganie powstawaniu zmian zwyrodnieniowych stawów,
– prewencja powikłań neurologicznych pod postacią głuchoty i innych objawów ze strony nerwów czaszkowych oraz zmian o typie stenozy kanałowej w przypadku zajęcia kręgosłupa,
– poprawa jakości życia pacjentów, choć nie należy tego wskazania uznawać za jedyne ani pierwszoplanowe w podejmowaniu leczenia, to ma ono istotne znaczenie w ocenie skuteczności terapii,
– planowe leczenie operacyjne,
– zapobieganie transformacji nowotworowej ognisk patologicznych.
Na liście leków zarejestrowanych przez amerykański Urząd ds. Żywności i Leków ( Food and Drug Administration, FDA) do leczenia choroby Pageta kości znajdują się [30]:
rizedronian – podawany doustnie w dawce 30 mg dziennie przez 2 miesiące
alendronian – podawany doustnie w dawce 40 mg dziennie przez 6 miesięcy
pamidronian – podawany dożylnie w ciągłym wlewie kroplowym w dawce 30 mg przez 4 godziny w trzech kolejnych dniach do łącznej dawki 90 mg (schemat podawania zaaprobowany przez FDA); częściej stosowanym sposobem podawania preparatu jest 2-4 godzinny wlew kroplowy 60 mg w dwóch kolejnych dniach do łącznej dawki 120 mg
tiludronian – podawany doustnie w dawce 400 mg przez 3 miesiące
etydronian – podawany doustnie w dawce 200 do 400 mg dziennie przez 6 miesięcy (zaleca się stosowanie wyższych dawek, co często wiąże się z występowaniem poważnych objawów niepożądanych ze strony przewodu pokarmowego)
kalcytonina łososiowa – podawana w postaci iniekcji w dawce 50-100 IU dziennie 3 razy w tygodniu przez 6-18 miesięcy (lek nie ma już praktycznie zastosowania klinicznego w tej jednostce chorobowej, gdyż uzyskiwana redukcja parametrów metabolicznych jest krótkotrwała).
Skuteczność leczenia pacjentów z chorobą Pageta za pomocą bisfosfonianów może być monitorowana nie tylko na podstawie oceny klinicznej oraz aktywności fosfatazy alkalicznej w surowicy krwi. Wykazano, że podawanie pamidronianu chorym z PD powoduje znaczny wzrost stężenia OPG, nieznamienne obniżenie stężenia RANKL oraz istotny spadek stosunku RANKL do OPG. U pacjentów bez poprawy klinicznej poziomy RANKL i OPG pozostawały natomiast na nie zmienionym poziomie [15]. Stwierdzono także, ze stosowanie rizedronianu pozwala uzyskać wzrost OPG u wszystkich leczonych pacjentów [16].
Sugeruje się, że działanie bisfosfonianów może być więc mediowane przez bezpśredni lub pośredni wpływ na układ RANKL I OPG [15].
Podjęcie decyzji o leczeniu operacyjnym w przypadku choroby Pageta kości jest trudne. Wymaga dobrej współpracy pomiędzy zespołem lekarskim i samym pacjentem. Chory powinien poznać wszystkie okoliczności przemawiające na korzyść interwencji chirurgicznej, ale również zagrożenia i konsekwencje ewentualnego odstąpienia od zabiegu. Choć jasne wydaje się, że takie postępowanie jest powszechnie obowiązującą doktryną medyczną, to w przypadku tej choroby ma znaczenie kluczowe.
Przeprowadzenie zabiegu operacyjnego w obrębie kości u osób z PD jest często trudne technicznie. Wynika to ze specyfiki samego schorzenia. Po pierwsze kość w przebiegu osteitis deformans ulega pogrubieniu i deformacji. Staje się ona twarda, przez co większość zabiegów stwarza znaczne trudności. Zaburzenie mikroarchitektury kości wiąże się również z powstaniem lakunarnych zbiorników naczyniowych w obrębie kości, które są miejscami nasilonego krwawienia śród- i pooperacyjnego. Ponadto kość o zmienionym metabolizmie może cechować się zmienionym tempem gojenia po zabiegach operacyjnych. Te fakty powodują, że większość dostępnych publikacji odnosi się do problemu leczenia chirurgicznego w przebiegu choroby Pageta kości z dużym dystansem. Selby i wsp. [33] wskazują, że przygotowanie farmakologiczne pacjentów do zabiegów operacyjnych ma na celu zmniejszenie krwawienia śródoperacyjnego i ograniczenie ryzyka dalszych deformacji. Autorzy uważają, że warunkiem konicznym do spełnienia przed zabiegiem operacyjnym jest obniżenie aktywności fosfatazy alkalicznej o co najmniej 25% w stosunku do najwyższej zanotowanej wartości.
Choroba Pageta kości jest schorzeniem o złożonej etiopatogenezie, zmiennej i zróżnicowanej symptomatologii. Wskazania do podjęcia leczenia nie są jednoznacznie ustalone i dlatego cennym i celowym jest stworzenie ośrodków specjalizujących się w tym zagadnieniu. Wpłynie to przede wszystkim na zwiększenie skuteczności diagnostycznej przy jednoczesnej redukcji kosztów tych działań oraz poprawi efektywność leczenia.
Nadzieje, jakie wiąże się z wprowadzeniem na rynek kolejnych leków o wysokiej skuteczności w zakresie wpływu na zaburzony w przebiegu choroby metabolizm kostny, są wielkie. Zoledronian, kolejny spośród bisfosfonianów, który uzyska rejestrację do leczenia choroby Pageta kości, wydaje się bardziej skuteczny, ale jednocześnie droższy niż powszechnie stosowane pamidronian, klodronian czy rizedronian. Piśmiennictwo
1. Tiegs RD: Paget´s disease of bone: indications for treatment and goals of therapy; Clin Ther 1997;19(6):1309-1329.
2. http://www.paget.org
3. Rousiere M, et al.: Paget´s disease of bone; Best Prac Res Clin Rheum 2003;17(6):1019-41.
4. Ankron MA, Shapiro JR: Paget´s disease of bone. J Am Ger Soc 1998;46(2)1025-26.
5. Barker DJP: The epidemiology of Paget´s disease of bone. Br Med Bull 1984;40(4):396-400.
6. Cooper C, et al.: The epidemiology of Paget´s disease in Britain: Is the prevalence decreasing? J Bone Miner Res 1999;14:192-197.
7. http://www.paget.org.uk
8. The Merck Manual of Diagnosis and Therapy, Section 5, Chapter 58; http://www.merck.com /mrkshared/mmanual/homejsp
9. Rebel A, et al.: Is Paget´s disease of bone a viral infection ? Calcif Tissue Res 1977;22(Supl):283-6.
10. Singer FR: Update on the viral etiology of Paget´s disease of bone. J Bone Miner Res 1999;14(suppl 2): 29-33.
11. Basle M, et al.: Demonstration by immunofluorescence and immunoperoxidase of an antygen of measles type in osteoclasts of Paget´s disease of bone. Bull Assoc Anat 1979;63:263-72.
12. Mills BG, et al.: Evidence for both respiratory syncytial wirus and measles virus antigens in the osteoclasts of patients with Paget´s disease of bone; Clin Orthop 1984;183:303-11.
13. Resnick D, Niwayama G: Diagnosis of bone and joint disorders. II edition, Saunders, Philadeplhia 1988.
14. Smith R: Paget´s disease of bone: Past and Present, Bone 1999;24(5):1S-2S.
15. Martini G, et al.: Serum OPG and RANKL levels before and after intravenous bisphosphonate treatment in Paget´s disease of bone.Bone. 2007 Feb;40(2):457-63.
16. Mossetti G, et al.: Interleukin-6 and osteoprotegerin systems in Paget´s disease of bone: relationship to risedronate treatment.Bone. 2005 Mar;36(3):549-54.
17. Sun SG, et al.: Bone stromal cells in pagetic bone and Paget´s sarcoma express RANKL and support human osteoclast formation.J Pathol. 2006 May;209(1):114-20.
18. Menaa C, et al.: 1,25-Dihydroxyvitamin D3 hypersensitivity of osteoclast precursors from patients with Paget´s disease. J Bone Miner Res. 2000 Feb;15(2):228-36.
19. Neale SD, et al.: Osteoclast differentiation from circulating mononuclear precursors in Paget´s disease is hypersensitive to 1,25-dihydroxyvitamin D3 and RANKL. Bone. 2000;27: 409-416.
20. Kurihara N, et al.: Role of TAFII-17, a VDR binding protein, in the increased osteoclast formation in Paget´s disease. J. Bone Miner. Res. 2004;19:1154-1164.
21. Laurin N, et al.: Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 2002;70:1582-1588.
22. Hocking LJ, et al.: Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget´s disease. Hum. Mol. Genet. 2002;11:2735-2739.
23. Daroszewska A, Ralston SH: Mechanisms of disease: genetics of Paget´s disease of bone and related disorders. Nat Clin Pract Rheumatol. 2006 May;2(5):270-7.
24. Sanz L, et al.: The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 1999;18:3044-3053.
25. Sanz L, et al.: The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576-1586.
26. Kurihara N, et al.: Mutation of the sequestosome 1 ( p62) gene increases osteoclastogenesis but does not induce Paget disease. J Clin Invest. 2007 January 2; 117(1): 133-142.
27. Johnson-Pais TL: Three novel mutations in SQSTM1 identified in familial Paget´s disease of bone. J. Bone Miner. Res. 2003;18:1748-1753.
28. Falchetti A, et al.: Segregation of a M404V mutation of the p62/sequestosome 1 (p62/SQSTM1) gene with polyostotic Paget´s disease of bone in an Italian family. Arthritis Res. Ther. 2005;7:R1289-R1295.
29. Singer FR, et al.: Absence of evidence of Paget´s disease of bone in subjects who harbor sequestosome 1 mutations [abstract]. J. Bone Miner. Res. 2004;19:M520.
30. Schneider D, Hofmann MT, Peterson JA: Diagnosis and Treatment of Paget´s Disease of Bone; Amer Fam Phys. 2002, 65(10):2069-72.
31. Kanis JA: Pathophysiology and Treatment of Paget´s Disease of Bone. 2nd Ed. London: Martin Dunitz; 10; 1998.
32. Borejko M, Dziak A: Badanie radiologiczne w ortopedii, PZWL 1973 (wyd. 1).
33 Selby PL, et al.: Guidelines on the Management of Paget´s Disease of Bone, Bone 2002;31(3):10-19.
34. Meunier PJ, Vignot E: Therapeutic Strategy in Paget´s Disease of Bone. Bone 1995; 17(5): 489-491.
35. Tiegs R: Paget´s disease of bone: Indications for treatment and goals of therapy. Clin Ther 1997;19(6):1309-1329.
otrzymano/received: 2008-03-26 zaakceptowano/accepted: 2008-05-26 Adres/address: *Dariusz Chmielewski Szpital HOSPITEN - Lanzarote, Puerto del Carmen/Hiszpania Lomo Gordo s/n, 35510 Puerto del Carmen/Lanzarote, Espana e-mail: dariusz.chmielewski@yahoo.es The whole paper Choroba Pageta kości is also available at On-line Medical Library. |
  Please click on selected cover
 |






Chcesz być na bieżąco? Polub nas na Facebooku: strona Wydawnictwa na Facebooku |