|
© Borgis - Postępy Nauk Medycznych 5, s. 268-278
*Alicja Nauman, Agnieszka Piekiełko-Witkowska, Olga Turowska, Piotr Popławski, Adam Master, Zbigniew Tański1, Joanna Lampkowska, Anna Wójcicka, Izabela Brózda, Monika Puzianowska-Kuźnicka
Zaburzenia szlaków sygnałowych hormonu tarczycy – trijodotyroniny – w raku nerki typu jasnokomórkowego
Disturbances of thyroid hormone signal pathway – in clear cell renal cell carcinoma
Zakład Biochemii i Biologii Molekularnej Centrum Medycznego Kształcenia Podyplomowego w Warszawie
Kierownik Zakładu: prof. dr hab. med. Andrzej Gardas 1Oddział Urologii, Szpital Specjalistyczny, Ostrołęka Ordynator: dr med. Zbigniew Tański Streszczenie
Praca przedstawia wyniki badań prowadzone przez Zespół od ponad 15 lat nad udziałem hormonu tarczycy – trijodotyroniny (T3) i jej receptorów jądrowych (TR) w raku nerkowo-komórkowym – w jego najczęstszej postaci – raka zbudowanego z komórek jasnych (ccRCC). Na podstawie naszych początkowych wyników badań postawiliśmy hipotezę, że T3 może być związana z nowotworzeniem. T3 jest czynnikiem niezbędnym dla prawidłowej kontroli podstawowych procesów komórkowych takich jak podział komórkowy, apoptoza czy różnicowanie komórek. Wszystkie te procesy zależne są nie tylko od stężenia i biodostępności T3, ale również od rodzaju, ilości i aktywności TR oraz od obecności innych czynników, z którymi TR oddziałują na poziomie molekularnym. Udowodniliśmy, że zmiany w ekspresji genów TR, prowadzą do zaburzeń przekazywania sygnału od T3 do genów docelowych, w sposób zależny od obecności T3 (hamowanie bez liganda, aktywacja w obecności liganda). Udowodniliśmy, że T3 może wywierać swój efekt biologiczny tylko wtedy, gdy TR posiada prawidłową budowę. Słowa kluczowe: hormon tarczycy, trijodotyronina, receptory jądrowe, ekspresja genów zależnych od trijodotyroniny, rak nerki jasnokomórkowy
Summary
The article describes the results of more that 15 years research on the role of thyroid hormone, triiodothyronine (T3) and its nuclear receptors in clear cell renal cell carcinoma (ccRCC). On the basis of our primary research we made a hypothesis that T3 can be engaged in neogenesis. T3 is indispensable for the proper control of the basic cellular processes such as cell division, apoptosis and differentiation. All these processes are dependent not only on concentration and bioavailability of T3 but also on type, amount and activity of TRs and the presence of other TRs molecules. We proved that the changes in TR expression lead to disturbances in signal transduction from T3 to the T3 regulated genes. The disturbances are T3 dependent (the expression is lowered in the absence of T3 and activated in the presence of T3). We also proved that T3 can exert its effect only in the presence of TRs which possess proper sequence. Key words: thyroid hormone, triiodothyronine, nuclear receptors, T3 dependent gene expression, clear cell renal cell carcinoma
Wprowadzenie
Szlaki sygnałowe, poprzez które działa hormon tarczycy, trijodotyronina (T3), i ich wzajemne połączenia tworzą sieć przenoszącą informację zapisaną w strukturze chemicznej hormonu, doprowadzając do swoistej odpowiedzi na poziomie genomu. Specyficzna odpowiedź osiągana jest dzięki istnieniu jądrowych białek receptorowych, z którymi T3 się wiąże. Receptory T3 ( Thyroid hormone Receptor, TR) są zależnymi od liganda czynnikami transkrypcyjnymi, które wiążą się ze specyficznymi sekwencjami, TRE ( Thyroid Responsive Element) (1), w obrębie części regulatorowych genów docelowych.
Trijodotyronina jest czynnikiem niezbędnym dla prawidłowej kontroli podstawowych procesów komórkowych, takich, jak proliferacja komórki (pro- lub anty-proliferacyjne działanie T3 zależne jest w znacznym stopniu od rodzaju tkanki i etapu rozwojowego), śmierć fizjologiczna komórki (apoptoza) oraz różnicowanie komórek. Równowaga pomiędzy tymi procesami jest konieczna dla prawidłowej kontroli szybkości podziałów komórkowych i utrzymania homeostazy organizmu. Wszystkie te procesy zależne są od stężenia i biodostępności T3, rodzaju, ilości i aktywności TR oraz od obecności innych czynników, z którymi TR oddziałują na poziomie molekularnym. Z licznych badań, prowadzonych między innymi w naszym zespole wynika, że zmiany w ekspresji genów TR prowadzą do zaburzeń w funkcjonowaniu komórki, tkanki i całego organizmu. Prawdopodobne jest więc, że w nowotworach, na skutek zmian dotyczących TR, dochodzi do zaburzeń przekazywania sygnału od T3 do genów docelowych.
W prowadzonych przez nas od 1990 roku badaniach nad udziałem T3 i jej receptorów w raku nerki typu jasnokomórkowego ( clear cell Renal Cell Carcinoma ccRCC) zajmujemy się przede wszystkim identyfikacją genów o zaburzonej ekspresji wywołanej zmianą stężenia biodostępnej T3 lub nieprawidłowościami dotyczącymi TR w komórce docelowej. W komórkach nowotworowych nerki analizowaliśmy regulację ekspresji genów będących pod ścisłą kontrolą T3, a mianowicie genów kodujących białka receptorowe TR i genu jodotyroninowej dejodynazy typu I (enzymu kontrolującego poziom T3 w komórce). Ponadto, badaliśmy ekspresję genów kodujących białka kontrolujące przejście z fazy G1 do fazy S cyklu komórkowego (cykliny E, czynniki transkrypcyjne rodziny E2F1-8, geny białek rodziny retinoblastoma (Rb, p107, p130), kinazę cdk2), których ekspresja jest również zależna od T3. Badania prowadzono w tkankach (guz nowotworowy nerki/tkanka nienowotworowa nerki) oraz w liniach komórkowych wywodzących się z raka nerki i ze zdrowych kanalików nerkowych (Caki-2, Caki-1 i HK2). Badając różnice w regulacji ekspresji genów, szczególną uwagę poświęcono zmianom na poziomie trans-kryptomu, czyli:
– regulacji pretranskrypcyjnej: regulacji poprzez wiązanie czynników transkrypcyjnych, np. TR, z regulatorowymi sekwencjami promotorowymi (TRE),
– regulacji potranskrypcyjnej: regulacji przez usuwanie intronów z pre-mRNA i różnicowe łączenie eksonów ( alternative splicing),
– regulacji pretranslacyjnej: regulatorowym sekwencjom w obrębie 5´ UTR (region niekodujący, UnTranslated Region) i 3´UTR.
Ponadto:
– geny o zróżnicowanej ekspresji identyfikowano również na podstawie obrazu rozdziału w żelach ich białkowych produktów,
– określano aktywację receptorow TR przez oznaczanie maksymalnego wiązania z T3
Analizowano 200 skrawków tkanek pobranych z guza nowotworowego, 200 skrawków tkanek nienowotworowych pobranych z przeciwnego bieguna tej samej nerki i 20 tkanek nienowotworowych, pobranych z nerek operowanych z innych powodów niż nowotwór. Stworzono bank cDNA, DNA, RNA oraz kolekcję wektorów zawierających sekwencje kodujące i niekodujące receptory jądrowe trijodotyroniny TRα i TRβ, gotowych do dalszej analizy. Przeprowadzono też genetyczną ocenę badanego materiału biologicznego pod kątem występowania niestabilności mikro-satelitarnej ( MicroSatellite Instability, MSI) oraz utraty heterozygotyczności ( Loss Of Heterozygosity, LOH). Wykazano, że w obrębie grupy nowotworów określonych histopatologicznie jako homogenna w aspekcie stopnia zróżnicowania (G1, G2, G3) znajdują się tkanki wykazujące różnice genetyczne w zakresie niestabilności mikrosatelitarnej i utraty heterozygotyczności. Analiza niestabilności mikrosatelitarnej wykazała obecność MSI w 12,50% analizowanych nowotworów (ryc. 1), zaś analiza utraty heterozygotyczności wykazała obecność LOH w 9,35% analizowanych tkanek ccRCC (ryc. 2).
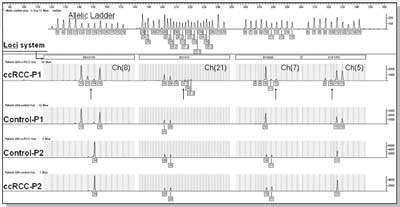 Ryc. 1. Chromatogramy genotypowania niestabilności mikrosatelitarnej-MSI. Porównanie dwóch przykładowych profili ccRCC i kontroli (P1-próbka z obserwowanym profilem MSI, P2-bez MSI). Poszczególne linie wynikowe zawierają informacje o kolejnych analizowanych systemach STR występujących na różnych chromosomach i oznaczonych: Ch(nr chromosomu). Strzałki wskazują zwielokrotnienie alleli w poszczególnych loci, wskazując na obecność MSI w przykładowej lini P1 ccRCC.
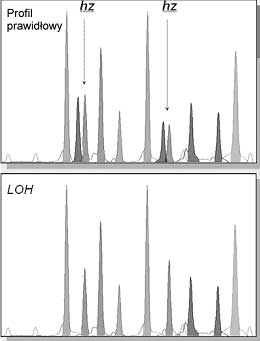 Ryc. 2. Chromatogramy genotypowania utraty heterozygotyczności – Porównanie przykładowych profili ccRCC i kontroli w badanych loci typu SNP, występujących na chromosomie 3, sekwencji genu THRB. „Profil prawidłowy” prezentuje genotyp próbki kontrolnej (bez obserwowanej utraty heterozygotyczności), na co wskazują zaznaczone strzałkami (hz) heterozygotyczne miejsca SNP (podwójne piki dwóch alleli w pojedynczym loci na dwóch różnych chromosomach homologicznych). „LOH” prezentuje profil próbki ccRCC z obserwowaną homozygotycznością we wszystkich badanych loci, co wskazuje na utratę heterozygotyczności w obrębie analizowanej sekwencji chromosomu trzeciego
Ekspresja jodotyroninowej dejodynazy w komórkach nowotworowych
Jodotyroninowe dejodynazy, które są oksydoreduktazami [EC.3.8.14] i należą do rodziny selenoprotein, do aktywacji których konieczna jest obecność zredukowanych związków tiolowych (RSH), pełnią kluczową rolę w homeostatycznym systemie kontrolującym stężenie hormonów tarczycy w każdej komórce. Odpowiadają za kontrolę prawidłowego rozwoju, wzrostu, różnicowania i specyficznego metabolizmu komórek. Działają w tyreocytach i we wszystkich komórkach pozatarczycowych, tworząc swoisty wewnątrzkomórkowy system enzymów błonowych, odpowiedzialnych nie tylko za biotransformację (konwersję) prohormonu, tyroksyny (T4), do aktywnego hormonu T3, ale również za specyficzną regulację poziomu T4 i T3 w każdej komórce. Synteza dejodynaz jest ściśle kontrolowana przez stężenie T3 w komórce, a stężenie T3 przez jodotyroninowe dejodynazy.
Wyróżnia się trzy typy jodotyroninowych dejodynaz: dejodynaza typu I (D1) dejodynaza typu II (D2) i dejodynaza typu III (D3). Jodotyroninowe dejodynazy katalizują reakcje odjodowania hormonów tarczycy (ryc. 3). Reakcja odjodowania T4 w pozycji 5´ ( Outer Ring Deiodination, ORD, odjodowanie pierścienia zewnętrznego, fenolowego), katalizowana przez dejodynazę typu I (5´D1) i typu II (5´D2) prowadzi do powstania aktywnej formy hormonu, T3. Reakcja odjodowania T4 w pozycji 5 ( Inner Ring Deiodination, IRD, odjodowanie pierścienia wewnętrznego, tyrozylowego), katalizowana przez dejodynazę typu I (5D1) i typu III (5D3), prowadzi do powstania nieaktywnej pochodnej, tzw. rewers trijodotyroniny (rT3). Zarówno trijodotyronina, jak i rT3, podlegają dalszym reakcjom odjodowania z wytworzeniem nieaktywnych dijodotyronin (T2).
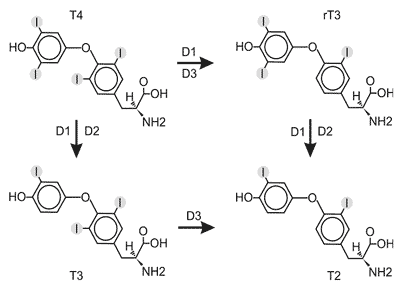 Ryc. 3. Reakcje katalizowane przez jodotyrononowe dejodynazy typu: 1 (D1), 2 (D2) i 3 (D3). T4: 3,5,3´5´-tetrajodotyronina (tyroksyna), T3: 3,5,3´- trijodotyronina, rT3: 3´5´3- rewers-trijodotyronina, T2: 3,3´-dijodotyronina.
Dejodynazy są zakotwiczonymi w błonach komórkowych białkami o masie 29-33 kDa. D1 i D3 lokalizują się w błonie cytoplazmatycznej, natomiast D2 zakotwiczona jest w błonie retikulum endoplazmatycznego (2,3). Funkcjonują prawdo-podobnie jako dimery (4,5). Centrum aktywne dejodynaz zawiera nietypowy aminokwas, selenocysteinę (Sec), kodowany przez trójkę nukleotydów UGA, w większości przypadków traktowanych w procesie translacji jako kodon STOP (6-8). Podczas syntezy łańcucha polipeptydowego kompleks translacyjny rozpoznaje kodon UGA jako kodujący Sec-tRNA dzięki współdziałaniu elementów trans (białek SBP2 i EF-sec) oraz elementu cis – zlokalizowanej w rejonie 3´UTR transkryptu dejodynazy sekwencji o nazwie SECIS, tworzącej strukturę tzw. szpilki do włosów (9).
Jodotyroninowa dejodynaza typu I
Dejodynaza typu I była pierwszym odkrytym enzymem katalizującym konwersję T4 do T3 (10,11), pierwsza też została sklonowana (6, 12). D1 jako jedyna spośród dejodynaz, katalizuje reakcję odjodowania w pozycji 5´ (z wytworzeniem T3 i T2), jak i w pozycji 5 (z wytworzeniem rT3). D1 wykazuje najwyższe powino-wactwo do rT3, mniejsze do T4 i T3 (rT3< W stanie eutyreozy D1 odpowiada za syntezę około 34% obwodowej T3, natomiast w stanie tyreotoksykozy jej udział wzrasta do 67% (i więcej) w zależności od stopnia zaawansowania choroby (16). Aktywność D1 specyficznie hamuje 6-n-propylotiouracyl (PTU); właściwość ta jest wykorzystywana do różnicowania aktywności D1 od pozostałych dejodynaz.
Gen kodujący D1 ( Dio1) w genomie ludzkim zlokalizowany jest w pozycji 1p32-33, składa się z 4 eksonów (17). Transkrypcja Dio1jest regulowana między innymi przez hormony tarczycy. W promotorze Dio1znajdują się dwa pozytywne TRE: klasycznyTRE-DR4 (18,19) oraz nietypowy TRE-DR10 (18), umożliwiające aktywację transkrypcji genu przez receptory TR, a także miejsca wiązania czynnika transkrypcyjnego Sp1 (19, 20).
Pierwotny transkrypt Dio1podlega procesowi różnicowego składania eksonów, w wyniku którego powstają warianty transkrypcyjne o różnej sekwencji, w tym pozbawione elementów kodujących rejon białka zawierający centrum aktywne (ryc. 4).
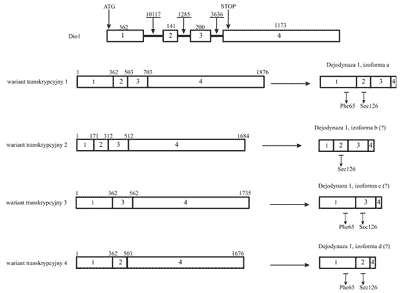 Ryc. 4. Gen, transkrypt i produkt białkowy ludzkiej dejodynazy 1. Rycina przedstawia cztery istniejące w bazie danych Entrez-Gene warianty splicingowe i powstające na ich matrycy produkty białkowe. W literaturze opisano jedynie wariant 1 i odpowiadającą mu izoformę a. Znakiem "?” oznaczono izoformy dejodynazy, których istnienie in vivo nie zostało jeszcze potwierdzone. Eksony zaznaczono prostokątami i ponumerowano od 1 do 4. Liczby ponad eksonami i intronami wskazują na ich wielkość w parach zasad. Na schemacie genu zaznaczono pozycje kodonów ATG i STOP. Na schemacie białka zaznaczono aminokwasy istotne z punktu widzenia aktywności enzymatycznej dejodynazy 1.
Ekspresja dejodynazy 1 w tkankach nowotworowych
Ekspresja genu Dio1jest zaburzona w nowotworach. Poziom mRNA i aktywności D1 są obniżone w raku brodawkowatym tarczycy (21-24), w raku jasnokomórkowym nerki (25, 26), w guzach glejowych mózgu (27), podwyższony w gruczolaku pęcherzykowym i raku pęcherzykowym tarczycy (21), raku piersi (28). Badania na liniach komórkowych raka piersi MCF-7 i MDA-MB-231 sugerują, że podwyższona ekspresja D1 ( Dio1) może stanowić marker różnicowania w komórkach nowotworowych (29).
Pierwsze doniesienia dotyczące zaburzonej ekspresji D1 w ccRCC zostały opublikowane przez nasz zespół w 1991 r. (25). Stwierdzono wówczas, że aktywność D1 była znacząco obniżona w tkance nowotworowej w porównaniu ze stanowiącą kontrolę zdrową tkanką otaczającą guz. Obniżona aktywność D1 korelowała z obniżonym poziomem białka. Kolejne badania wykazały znaczne zróżnicowanie aktywności D1 oraz jej ekspresji na poziomie mRNA pomiędzy próbkami kontrolnymi pobranymi od różnych pacjentów. Różnice w poziomie mRNA były nawet 10-krotne. W tkankach nowotworo-wych zarówno aktywność enzymatyczna jak i ilość mRNA D1 były prawie niewykrywalne (30, 26). Jedną z przyczyn obniżonej ekspresji D1 w nowotworach nerki może być zaburzona aktywacja transkrypcji Dio1przez receptory TR. Zmutowane wersje TR sklonowane z tkanek ccRCC aktywowały promotor genu Dio1znacznie słabiej, niż dzikie typy tych (31).
Analiza ekspresji Dio1na poziomie posttranskrypcyjnym
Analiza wariantów transkrypcyjnych klonowanych z tkanek ccRCC oraz z tkanek nienowotworowych z przeciwległego bieguna nerki nowotworowej, jak również z próbek pobranych z nerek nienowotworowych (powypadkowych, kamiczych, ektopowych, marskich) wykazała istnienie wielu nieopisanych do tej pory wariantów transkrypcyjnych Dio1, w tym wariantów pozbawionych eksonu 2, zawierającego rejon kodujący centrum aktywne. Zidentyfikowano m.in. warianty zawierające wszystkie 4 eksony, eksony: 1, 2, 4, eksony: 1 i 4, a nawet wariant składający się tylko z eksonu 1. Dokładna analiza poziomu ekspresji transkryptów genu Dio1,wykonana za pomocą techniki PCR czasu rzeczywistego ( real-time PCR) wykazała dramatycznie obniżony poziom ekspresji wszystkich wariantów transkrypcyjnych (o około 90%) w tkankach nowotworowych w porównaniu z tkankami kontrolnymi (32). Zaburzony był również proces różnicowego składania eksonów. W tkankach nowotworowych stwierdzono istotny statystycznie spadek stosunku wariantów transkrypcyjnych zawierających rejon kodujący centrum aktywne białka i wariantów pozbawionych tego rejonu. Stwierdzono, że zaburzenia różnicowego składania eksonów D1 wynikają prawdopodobnie z nieprawidłowej ekspresji czynników splicingowych SF2/ASF i hnRNPA1 w ccRCC (Piekiełko-Witkowska A., wyniki niepublikowane).
Analiza ekspresji receptorów jądrowych trijodotyroniny
Receptory jądrowe trijodotyroniny, TR, to średniej wielkości białka jądrowe (TRβ1- 461 aminokwasów; 53 kDa), kodowane przez geny zlokalizowane na chromosomach trzecim ( THRB - 3p24.2) i siedemnastym ( THRA - 17q23).
W procesie różnicowego składania pierwotnego transkryptu powstają po dwie główne izoformy każdego receptora – odpowiednio TRα1 i TRα2 oraz TRβ1 i TRβ2. Receptory jądrowe T3 wpływają na ekspresję genów docelowych poprzez selektywne wiązanie się z promotorowymi sekwencjami DNA typu HRE ( Hormone Response Element), wzmacniając tym samym (lub osłabiając) wydajność transkrypcji. TR, podobnie jak inne białka z rodziny receptorów jądrowych, wykazują obecność dobrze scharakteryzowanych domen funkcjonalnych (ryc. 5). Wiązanie hormonu powoduje zmiany konformacyjne białka powodując zmiany w oddziaływaniu TR z koaktywatorami i korepresorami.
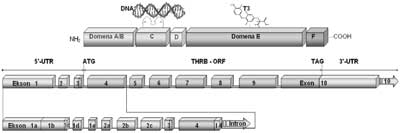 Ryc. 5. Schemat budowy domenowej białka receptorowego dla trijodotyroniny TRβ1 (A.) oraz układu eksonów transkryptu THRB (B.).
W budowie receptora TRβ1 wyróżniono: domenę A/B, odpowiedzialną za wiązanie białek regulatorowych oraz niezależną od T3 aktywację transkrypcji, domenę C wiążącą DNA (8Cys=2 palce cynkowe), domenę łącznikową D zawierającą sygnał lokalizacji jądrowej, domenę E odpowiedzialną za wiązanie hormonu (T3) i dimeryzację receptorów oraz domenę F odpowiedzialną za zależną od T3 aktywację transkrypcyjną receptora. Na schemacie przedstawiono ponumerowane kolejno eksony transkryptu THRB (B.) z wyszczególnieniem eksonów 5´UTR (1a, 1b, 1c, 1d, 1e, 2a, 2b, 2c) powstających w procesie różnicowego składania pierwotnego transkryptu do alternatywnych form mRNA, różniących się sekwencją obszaru regulatorowego nie ulegającego translacji. Na schemacie zaznaczono również kodon startu translacji, kodon zamknięcia ramki odczytu (TAG) oraz sekwencję kodującą (THRB-ORF). Regulacja pretranskrypcyjna ekspresji TR w jasnokomórkowym raku nerki: regulacja poprzez wiązanie czynników transkrypcyjnych - receptorów trijodotyroniny (TR) z regulatorAmi sekwencjami promotorowymi (TRE)
W pracy dotyczącej ekspresji TR w raku jasnokomórkowym nerki (33) stwierdzono, że ilość mRNA TRα1 była znamiennie zmniejszona w większości przypadków ccRCC w porównaniu z odpowiadającymi im tkankami kontrolnymi, natomiast ekspresja TRβ1 na poziomie mRNA była bardziej heterogenna: w 70% raków była znacząco zmniejszona, zaś w 30% – zwiększona w guzach w porównaniu z odpowiadającymi im kontrolami. Średnia ilość białka TRα1 była wyższa w ccRCC w porównaniu z tkankami kontrolnymi, podczas, gdy średnia ekspresja TRβ1 na poziomie białka była obniżona w tkankach tych samych nowotworów. Zwiększenie ilości TR niekoniecznie przekładało się na zwiększenie aktywności genów docelowych trijodotyroniny, ponieważ funkcja TR może być, i zwykle jest, zaburzona wskutek różnych mechanizmów, na przykład mutacji w genach kodujących TR, albo, prawdopodobnie, zaburzeń w modyfikacjach potranslacyjnych białka. W ccRCC znaleziono mutacje w receptorze trijodotyroniny (34). Mutacje dotyczyły obu izoform TRα1 i TRβ1; większość z nich znajdowała się w domenie wiążącej ligand, kilka zaś w domenie wiążącej DNA. Ponadto, odsetek tkanek ccRCC, w których stwierdzono mutacje TR wzrastał wraz ze spadkiem zróżnicowania histologicznego nowotworu (34). Badania funkcjonalne in vitro mutantów TR wykazały, że w zależności od miejsca, gdzie doszło do zmiany aminokwasu, mutanty takie nieprawidłowo wiążą T3 lub/i DNA oraz w sposób niedostateczny aktywują geny docelowe (34-38).
Regulacja ekspresji THRB na poziomie potranskrypcyjnym
Składanie pre-mRNA ( splicing) THRB (ryc. 5B) polega na usuwaniu z pierwotnego transkryptu preRNA sekwencji niekodujących (intronów) i łączeniu pozostałych eksonów w gotowy do translacji mRNA. W wyniku alternatywnego składania mogą powstawać kodowane przez ten sam gen białka, różniące się od siebie sekwencją aminokwasów i funkcją, jak również białka identyczne, chociaż kodowane przez mRNA różniące się sekwencją UTR ( UnTranslated Region). UTR to część mRNA nie ulegająca translacji, położona w kierunku 5´ (5´-UTR) lub 3´ (3´-UTR) od sekwencji kodującej. Procesowi składania pre-mRNA podlegają również wspomniane obszary mRNA (5´- oraz 3´-UTR), których funkcja w dojrzałym mRNA polega na regulacji ekspresji genu na poziomie pre-translacyjnym. Najnowsze badania wskazują, że poziom translacji wariantów TR może być regulowany przez takie rejony mRNA (39). Innymi słowy, ilość powstającego białka TR jest zależna od wariantu splicingowego regionu 5´UTR mRNA. Obszary mRNA nie ulegające translacji mogą wpływać na trwałość cząsteczki RNA jak również inicjację oraz efektywność przebiegu samej translacji przez oddziaływanie obszarów 5´UTR na sekwencję kodującą, na tworzenie struktur II-rzędowych utrudniających inicjację translacji (40-41) jak również poprzez wiązanie małych cząsteczek regulatorowych miRNA do sekwencji 3´UTR, obniżających najczęściej efektywność procesu translacji (42-46). W ostatnim czasie wskazuje się na rolę miRNA oraz sekwencji 3´UTR w patogenezie nowotworów (46-48). Sekwencje mRNA nie ulegające translacji stanowią część znaczącego mechanizmu regulacji ekspresji genów na poziomie pre-translacyjnym.
Analiza ekspresji produktów alternatywnego składania THRB
Badania miały na celu określenie struktury i funkcji produktów różnicowego składania niekodujących sekwencji UTR pierwotnego transkryptu receptora TRβ1 w aspekcie jego ekspresji w prawidłowych i nowotworowo zmienionych komórkach. Analizowano ekspresję alternatywnie składanych form transkryptu izoformy receptora jądrowego trijodotyroniny TRβ1 w obrębie sekwencji nie ulegających translacji 5´-UTR. Ustalenie profilu form 5´-UTR wykonano w oparciu o łańcuchową reakcję polimerazy, z wykorzystaniem par starterów flankujących sekwencję niekodującą 5´UTR (w kierunku „forward”): ekson 1a,1b,1c lub 2a i sekwencję kodującą (w kierunku „reverse”): ekson 4 (początek ramki odczytu) lub ekson 10 (koniec ramki odczytu). Analizie jakościowej poddano pary kontrola-ccRCC, ustalone linie komórkowe różnego pochodzenia. Caki-2 (ccRCC), Caki-1 (przerzut skórny ccRCC), HK2 (kanaliki proksymalne nerki ludzkiej), HEK 293 (ludzkie komórki embrionalne nerki), MCF-7 (rak piersi), Jurkat cell line (ludzkie komórki limfoblastyczne T), Raji cell line (chłoniak B-komórkowy) jak również prawidłowe komórki nabłonka jamy ustnej oraz prawidłowe komórki jądrzaste krwi.
Analiza profili form 5´-UTR THRB wykazała, że: ekspresja alternatywnie składanych form splicingowych sekwencji 5´UTR THRB jest zależna od rodzaju tkanki i zaburzona w ccRCC. Prowadzi to do powstania różnych profili 5´UTR w ccRCC i kontrolach. Analiza profili 5´UTR wykazała zaburzenia alternatywnego splicingu nie tylko w ccRCC, ale również w ich kontrolach z przeciwległego bieguna nerki (różne profile eksonów 1a, 1b, 1c 5´UTR między kontrolami), co można próbować tłumaczyć parakrynnym i endokrynnym oddziaływaniem komórek ccRCC na tkankę otaczającą guz.
Analiza porównawcza profili wykazała, że w przeciwieństwie do wszystkich badanych eksonów THRB (1a, 1b, 1c), ekson 2a charakteryzuje się wysoką stabilnością profilu (układu prążków) nie tylko między różnymi tkankami, ale również między ccRCC oraz ich kontrolami, sugerując duże znaczenie tego eksonu dla ekspresji białka, jak również stabilność systemu różnicowego składania pierwotnego transkryptu dla eksonu 2 genu THRB.
Wykonano oznaczenia mRNA metodą PCR w czasie rzeczywistym ( real-time PCR) i stwierdzono znaczące obniżenie ilości mRNA receptorów trijodotyroniny w ccRCC w porównaniu z tkankami nienowo-tworowymi. W zakresie badań ilościowych 5´UTR wykonano analizę porównawczą real-time PCR mRNA eksonu 2a, 2b, 2c oraz transkryptu kodującego wyłącznie fragment białka TRβ1 (terminator i stop kodon znajdują się w transkrybowanej sekwencji intronu 4). Pomiar ilościowy analizowanych form 5´-UTR wykazał, że: w ccRCC dochodzi do znacznego obniżenia poziomu ekspresji wszystkich analizowanych form 5´UTR THRB w porównaniu do kontroli, wyizolowanych z przeciwległego bieguna nerki nowotworowej, co jest zgodne z wcześniejszą obserwacją znacznego obniżenia poziomu kodującej części THRB w ccRCC. Średnie stosunków (kontrola/ccRCC) wykazały, że poziom ekspresji badanych form 5´UTR eksonu 2a, 2b, 2c oraz formy 2a-Intron4 (skrócona forma receptora TR) różni się między ccRCC oraz ich kontrolami (ryc. 6). Wizualizacja żelowa amplikonów powstających form mRNA w parach ccRCC: kontrola wskazuje na zaburzenie ekspresji profilu powstających wariantów splicingowych 5´UTR w ccRCC. (50)
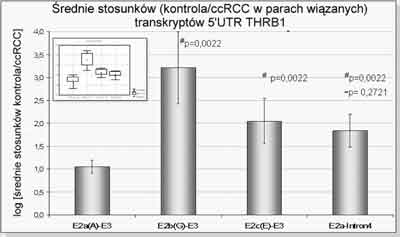 Ryc. 6. Ilościowy pomiar poziomu ekspresji produktów różnicowego składania THRB, zawierających ekson 2a, 2b lub 2c. Poszczególne słupki reprezentują kolejno średnie stosunków par wiązanych (kontrola/ccRCC) ekspresji alternatywnych form 5´UTR: transkryptów zawierających ekson 2a [E2a(A)-E3] wchodzący w skład formy A THRB (39), ekson 2b [E2b(G)-E3] wchodzący w skład formy G, ekson 2c [E2c(E)-E3] będący składową formy E jak również transkryptów zawierających ekson 2a i kodujących skrócone białko THRB-Intron4 [E2a-Intron4] (39). #różnice statystycznie istotne wyznaczone w oparciu o nieparametryczny test par wiązanych Wilcoxona przy założonym poziomie ufności <0,001.
Zaburzenia ekspresji cykliny E i jej niskocząsteczkowych izoform (LMW) w raku jasnokomórkowym nerki
Zaburzenia w regulacji przejścia z fazy G1 do S cyklu komórkowego są podstawową przyczyną onkogenezy. Ekspresja cykliny E, jednego z kluczowych białek regulujących przejście z fazy G1 do S, jest regulowana przez czynniki transkrypcyjne E2F. Transkrypcja genu kodującego E2F jest negatywnie regulowana przez jądrowe receptory trijodotyroniny. Wykazano zaburzoną ekspresję cykliny E i jej niskocząsteczkowych izoform ( Low Molecular Weight, LMW) w wielu nowotworach (51, 52).
W badaniach wykorzystano 26 skrawków tkanek nowotworowych (ccRCC), 26 skrawków tkanek nienowotworowych, pochodzacych z drugiego bieguna nerki operowanej (kontrola wewnętrzna) i 15 skrawków z tkanek pobranych z nerek operowanych z innego powodu niż nowotwór. Ekspresję cykliny E i jej izoform LMW oznaczano metodą Western-blott. Maksymalne wiązanie T3 do receptorów jądrowych (Bmax) oznaczano metodą radioreceptorową Scatcharda.
Analiza całkowitej ekspresji cykliny E wykazała statystycznie znamienne różnice pomiędzy zdrowymi kontrolami i tkankami nienowotworowymi. Poza tym, w tkankach nowotworowych wykazano znacząco podwyższoną ekspresję izoform LMW. Analiza Scatcharda wykazała istotnie obniżone Bmax w tkankach nowotworowych. Wykazano, że w grupie tkanek nowotworowych z Bmax obniżonym poniżej 25% występuje znacząco podwyższona ekspresja cykliny E (52). Zatem zaburzone wiązanie T3 z TR w ccRCC ma wpływ na wzrost ekspresji cykliny E i jej LMW, ekspresja tych białek jest negatywnie regulowana przez zależny od T3 i TR gen E2F1, którego produkt białkowy reguluje z kolei ekspresję cykliny E (ryc. 7). Zatem obniżone wiązanie T3 z TR powoduje zwiększoną ekspresję cykliny E, skrócenie fazy G1 i przyspieszoną proliferację. Powyższe wyniki badań wspierają hipotezę, że upośledzona funkcja T3/TR może być odpowiedzialna za proces nowotworzenia (52).
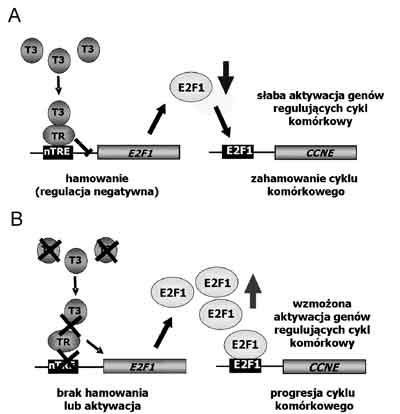 Ryc. 7. Schemat oddziaływań pomiędzy T3, TR, E2F1 i cykliną E
A) w tkankach zdrowych B) w tkankach nowotworowych. Zależności pomiędzy ekspresją jądrowych receptorów trijodotyroniny a ekspresją czynnika transkrypcyjnego E2F1 i cykliny E w raku jasnokomórkowym nerki
Wskutek zaburzeń dotyczących zarówno ilości biodostępnego hormonu, jak i ilości i funkcji TR, w tkankach nowotworowych może dojść do nieprawidłowej kontroli ekspresji genów docelowych T3, które biorą udział w regulacji proliferacji i apoptozy. W grupie tej znajduje się gen E2F1, kodujący kluczowy dla przebiegu cyklu komórkowego czynnik transkrypcyjny. E2F1 jest czynnikiem transkrypcyjnym, który nasila proliferację komórek na drodze aktywacji transkrypcji genów odpowiedzialnych za postęp cyklu komórkowego przez punkt restrykcyjny (R), znajdujący się na granicy faz G1 i S. W promotorze genu E2F1 stwierdzono obecność miejsca wiązania jądrowych receptorów trijodotyroniny, tzw. negatywnego miejsca odpowiedzi na hormony tarczycy (nTRE), za pośrednictwem którego kompleks T3 i TR hamuje aktywność tego genu. Z kolei w promotorze genu CCNE, kodującego cyklinę E, czynnik kluczowy dla postępu cyklu komórkowego, znajdują się miejsca wiązania czynników transkrypcyjnych E2F, zaś E2F1 jest silnym aktywatorem tego promotora. Z ostatnio opublikowa-nych prac wynika, że E2F1, najlepiej opisany członek rodziny E2F, może być zaangażowany w regulację transkrypcji aż 25-35% ludzkich genów (53). Cechą charakterystyczną E2F1, wyróżniającą ten czynnik na tle całej rodziny, jest posiadanie właściwości zarówno onkogenu, jak i supresora nowotworzenia (54) oraz spełnianie istotnej roli w procesie apoptozy (55-56). E2F1 pełni funkcję onkogenu poprzez nasilanie proliferacji komórek na drodze aktywacji transkrypcji genów odpowiedzialnych za postęp cyklu komórkowego, m.in. genu CCNE, CCNA, CDK2, CDK1, E2F1, E2F2, E2F3 (57-60). Wykazano, że nTRE, tzw. element Z, jest kluczowy dla regulacji transkrypcji E2F1oraz, że aktywność promotora E2F1 jest najwyższa w obecności bardzo niskich stężeń T3 i obniża się wraz ze wzrostem stężenia tego hormonu (61). W obecności niektórych dominujących-negatywnych mutantów TR promotor E2F1był nadmiernie aktywowany. Najnowsze badania tkanek ccRCC wykazały, że średnia ilość dwóch głównych, transkrypcyjnie aktywnych izoform TR (TRα1 i TRβ1) jest znamiennie podwyższona w porównaniu z obydwoma typami kontroli: fragmentami tych samych nerek, nieobjętymi nowotworem oraz nerkami nienowotworo-wymi. Stwierdzono, że podwyższeniu temu towarzyszy paradoksalny spadek maksymalnego wiązania T3 (Bmax) i wiązania DNA przez te receptory. Ponadto, w tkankach raka stwierdzono istotny statystycznie wzrost średniej ilości E2F1, zarówno na poziomie mRNA jak i białka. Nie wykazano natomiast istotnej statystycznie korelacji pomiędzy ilością białek TR, ich wiązaniem z DNA lub z T3, a ilością mRNA E2F1 we wszystkich grupach badanych tkanek. Obserwowano istotną statystycznie korelację pomiędzy ilością mRNA E2F1 i ilością białka E2F1 w tkankach ccRCC i w obydwu typach kontroli. W tkankach raka stwierdzono istotny statystycznie wzrost ilości mRNA cykliny E w porównaniu z tkankami kontrolnymi. Stwierdzono istotną statystycznie, dodatnią korelację pomiędzy ilością białka E2F1 i ilością mRNA cykliny E w nerkach nienowotworowych i w tkankach kontrolnych pobranych z przeciwległego bieguna nerki, ale nie w tkankach pochodzących z ccRCC (61). Podsumowując, aktywność promotora E2F1 jest regulowana przez trijodotyroninę i jej jądrowe receptory. Kontrola ta może być zaburzona w nowotworach wskutek nieprawidłowości dotyczących ilości i funkcji TR (ryc. 7). Rak jasnokomórkowy nerki jest nowotworem charakteryzującym się nadekspresją zarówno TR, jak i czynnika transkrypcyjnego E2F1 oraz cykliny E. Brak istotnej statystycznie korelacji pomiędzy ilością TR i ilością mRNA E2F1 również w nerkach nienowotworowych, sugeruje, że regulacja ekspresji E2F1 przez T3 nie jest jedynym, ani najważniejszym mechanizmem kontroli poziomu E2F1.
Wpływ hormonów tarczycy na ekspresję białek regulujących punkt kontrolny G1/S cyklu komórkowego w liniach komórkowych wywodzących się z komórek raka jasnokomórkowego nerki i kanalików proksymalnych zdrowej nerki ludzkiej
W tkankach pochodzących z ccRCC podwyższony, w porównaniu z tkankami kontrolnymi, poziom biorącego udział w regulacji przejścia z fazy G1 do S cyklu komórkowego, czynnika transkrypcyjnego E2F1 i cykliny E1, (zwanej zwykle cykliną E) (52,61). Biorąc pod uwagę wyniki powyższych badań zasadne wydawało się sprawdzenie wpływu T3 na ekspresję czynników regulujących przejście z fazy G1 do S i będącą tego efektem szybkość proliferacji komórek. Badania przeprowadzono na liniach komórkowych: HK2, Caki-2, Caki-1. Wykazano, że T3 wpływa odmiennie na proliferację komórek zdrowych i nowotworowych, hamując podziały w linii nienowotworowej HK2 i stymulując je w liniach nowotworowych (Caki-1, Caki-2) (ryc. 8A) (62). Wynika to najprawdopodobniej z odmiennej regulacji przejścia z fazy G1 do S cyklu komórkowego – w liniach nowotworowych T3 powodowała zwiększenie liczby komórek wchodzących w fazę S, podczas gdy w linii HK2 liczba komórek spadała.
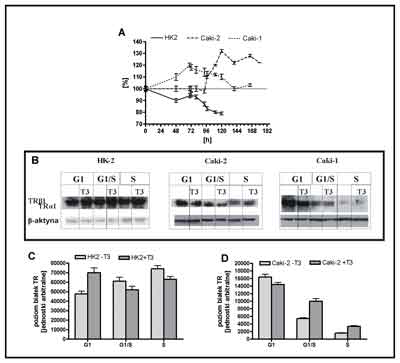 Ryc. 8. Wpływ trijodotyroniny na proliferacje komórek i ekspresję receptorów jądrowych trijodotyroniny
A) Wpływ trijodotyroniny na proliferację komórek linii HK2, Caki-2, Caki-1 B) Poziom TRβ1 i TRγα1 w lizacie komórkowym z badanych linii komórkowych hodowanych w obecności i przy braku 100 nM T3 na poszczególnych etapach cyklu komórkowego. Reprezentatywne immunobloty znakowane przeciwciałem rozpoznającym dwie główne izoformy receptora T3-TRβ1 i TRα1. C) Średni poziom TRβ1 i TRα1 w lizacie komórkowym z komórek linii HK2 hodowanych w obecności i przy braku 100 nM T3 na poszczególnych etapach cyklu komórkowego. D) Średni poziom TRβ1 i TRα1 w lizacie komórkowym z komórek linii Caki-2 hodowanych w obecności i przy braku 100 nM T3 na poszczególnych etapach cyklu komórkowego. Na szybkość proliferacji komórek wpływa szereg białek regulujących przejście z fazy G1 do S. Wykorzystując metodę Western blot i/lub SQ- Real time PCR wykonano analizę poziomu ekspresji czynników regulatorowych należących do trzech grup: i) aktywatorów proliferacji (czynników transkrypcyjnych E2F1-3, cyklin: E1, E2, kinazy białkowej cdk2), ii) inhibitorów proliferacji (czynników transkrypcyjnych E2F4-8, białek z rodziny retinoblastoma: Rb, p107, p130) oraz iii) jądrowych receptorów hormonów tarczycy, których wpływ na proliferację jest zróżnicowany. Poziom ekspresji mierzono w trzech etapach cyklu komórkowego (G1, G1/S i S). Wyniki tych badań wskazują, że hamowanie przez T3 proliferacji w linii HK2 wynika najprawdopodobniej z rosnącego pod wpływem T3 poziomu białka retinoblastoma (Rb), które tworząc kompleksy z czynnikami transkrypcyjnymi E2F1-3 uniemożliwia aktywację transkrypcji genów docelowych przez te czynniki. Wykazano też, że pod wpływem T3, w miarę progresji cyklu, następuje wzrost ekspresji hamujących proliferację kompleksów E2F5/p107 oraz czynników E2F6, E2F7, E2F8. Jednym z aktywatorów proliferacji jest kompleks cyklina E/cdk2. Inhibitorem działania tego kompleksu jest białko p107. W linii HK2 stwierdzono, że w miarę progresji cyklu następuje spadek poziomu ekspresji elementów składowych kompleksu aktywującego (cyklin E1 i E2, oraz kinazy białkowej cdk2) oraz wzrost ekspresji p107.
Aktywacja proliferacji przez T3 w liniach Caki-2 wynikała najprawdopodobniej z obniżenia poziomu ekspresji białek p107 i p130. Białka te tworzą, działające jako inhibitory proliferacji, kompleksy z czynnikami E2F4/5. W linii Caki-2 stwierdzono również obniżenie poziomu ekspresji elementów składowych kompleksu cyklina E/cdk2, będącego aktywatorem proliferacji. Obserwowany spadek poziomu białek kompleksu aktywującego mógł spowodować hamowanie proliferacji. Jednak, jak wspomniano wyżej, białka p107 i p130 są też inhibitorami działania kompleksów cyklinaE /cdk2. Efekt aktywacji proliferacji wskutek obniżenia poziomu ekspresji białek p107 i p130 był więc silniejszy niż hamowanie podziałów komórkowych wskutek obniżenia ekspresji elementów składowych kompleksu cyklina E/cdk2.
W linii Caki-1 za przyspieszenie proliferacji przez T3 odpowiadał prawdopodobnie wzrost poziomu ekspresji aktywującego podziały komórkowe czynnika transkrypcyjnego E2F1. W linii tej, wraz z progresją cyklu, następował też wzrost poziomu cykliny E2, należącej do grupy aktywatorów proliferacji.
T3 w różny sposób wpływała na poziom receptorów TR w linii HK2 i w liniach nowotworowych. W liniach nowotworowych (Caki-1, Caki-2) poziom receptorów był niższy niż w linii HK2, a w linii Caki-2, w odróżnieniu od linii HK2, spadał wraz z progresją cyklu (62) (ryc. 8).
W porównaniu z komórką prawidłową, w komórce nowotworowej ccRCC zależna od T3 regulacja podziału komórkowego jest istotnie zaburzona. Prawdopodobną przyczyną różnej odpowiedzi komórek na T3 są zaburzenia w regulacji ekspresji czynników regulujących przejście z fazy G1 do S. Otrzymane wyniki sugerują, że zaburzenia T3-zależnej kontroli proliferacji mogą być jednym z elementów prowadzących do transformacji nowotworowej w nerce.
Rak nerki wywodzi się z kanalika proksymalnego nerki, ma najczęściej postać litego guza i najczęściej jest zlokalizowany w korze nerki. Wskutek wysokiej ekspresji śródbłonkowego czynnika wzrostu naczyń (VEGF) (63, 64) nowotworowo zmieniona tkanka jest silnie unaczyniona, a wzrost guza następuje stosunkowo szybko. Nieprawidłowości genetyczne najczęściej występujące w ccRCC to utrata części lub całości krótkiego ramienia chromosomu 3, a także mutacje niektórych genów supresorowych. Utrata heterozygotyczności fragmentu chromosomu 3 (3p12-14, 3p21-22 lub 3p25-26) znajdowana jest w 98% tego typu raka (65, 66). U 57% chorych stwierdzono obecność mutacji genu supresorowego VHL, zlokalizowanego w regionie 3p25-26 (65). Spośród genów zlokalizowanych na ramieniu krótkim chromosomu 3, których ekspresja jest zaburzona, zidentyfikowano gen (3p24.2) receptora trijodotyroniny THRB (34,67).
U 34 chorych operowanych z powodu rozpoznanego ultrasonograficznie i/lub tomograficznie guza nerki oznaczono we krwi poziomy hormonów: T3, T4 i TSH. Pomiary stężeń tych hormonów przeprowadzono 1 dzień przed nefrektomią, 30 minut przed rozpoczęciem zabiegu i 30 minut po jego zakończeniu oraz w 1, 2, 5, 10 i 30 dniu po operacji. We wszystkich przypadkach stwierdzono zespół niskiej trijodotyroniny (68). Obserwowano korelację pomiędzy niskim stężeniem T3 i wolnej trijodotyroniny, a stopniem zaawansowania choroby nowotworowej. Najniższe stężenia trijodotyroniny obserwowano u chorych z nowotworem o najniższym stopniu zróżnicowania choroby podstawowej, i czasu jej trwania. Normalizacja poziomów T3 do wartości prawidłowych, zależała od stopnia zaawansowania choroby i ulegała normalizacji wraz z powrotem chorego do zdrowia (70). U najciężej chorych zespół niskiej T3 i T4, a więc istotny spadek stężenia we krwi nie tylko T3, ale i T4 był bardzo złym czynnikiem prognostycznym. Według De Groot´a (69), kiedy poziom całkowitej T4 we krwi spada do 2 μg/dL, prawdopodobieństwo zgonu pacjenta wynosi ok. 50%; przy poziomie poniżej 2 μg/dL prawdopodobieństwo zgonu osiąga 80%. (70). Problem czy zespoł chorób pozatarczycowych (NTIS, Non Thyroid Illness Syndrome) stanowi fizjologiczny mechanizm adaptacyjny ustroju mający na celu oszczędzanie energii w stanie jej zmniejszonej podaży lub wzmożonego zapotrze-bowania, czy też jest stanem spowodowanym podstawowym schorzeniem i prowadzi do pogłębienia zaburzeń, wynikających z choroby podstawowej (nowotworowej) jest do dzisiaj nierozstrzygnięty.
Podsumowanie
W świetle dotychczas otrzymanych wyników badań tkanek ccRCC ilość biodostępnej T3 jest obniżona, a niska aktywność transkrypcyjna genów THRA i THRB spowodowana jest nie tylko zaburzeniami ich struktury, zachodzącymi z różnych przyczyn na wielu poziomach regulacji ich ekspresji, ale również brakiem T3 w komórce docelowej. Świadczy o tym m.in. obniżony stopień maksymalnego wysycenia receptorów TR przez T3, czyli obniżone wiązanie TR z T3. Ponadto, obserwowany spadek ilości biodostępnej T3, może prowadzić, po pierwsze, do zwiększenia aktywacji promotorów genów z negatywnym nTRE. Wówczas receptor TR (nie związany z T3) aktywuje ekspresję tych genów, co powoduje zwiększenie transkrypcji i specyficznego mRNA (np. czynników transkrypcyjnych E2F1). Ponadto, spadek ilości T3 może prowadzić do obniżenia aktywacji promotorów z pozytywnym TRE (TR związane z T3), co w przypadku braku T3 prowadzi do obniżenia transkrypcji i spadku ilości specyficznego mRNA (np. genu Dio1 - dejodynazy typu1). Pierwotny transkrypt Dio1podlega procesowi różnicowego składania eksonów, w wyniku którego powstają warianty transkrypcyjne o różnej sekwencji, w tym pozbawione elementów kodujących rejon białka zawierający centrum aktywne, a więc pozbawione właściwości katalitycznych. Określenie struktury i funkcji poszczególnych produktów różnicowego składania eksonów THRB i powiązanie konkretnych sekwencji UTR ze zmianami w poziomie białka receptorowego wyjaśnia w pewnym stopniu uprzednio obserwowane przez nas zjawisko braku korelacji między poziomem mRNA i białka TR. Niezależnie jednak od specyficzności obserwowanych zaburzeń różnicowym składaniu sekwencji 5´UTR, identyfikacja struktury i funkcji niekodujących sekwencji UTR będzie miała znaczenie dla zrozumienia udziału receptorów TR w procesach nowotworzenia. Piśmiennictwo
1. Oetting A, Yen PM: New insights into thyroid hormone action. Best Pract Res Clin Endocrinol Metab 2007; 21: 193-208.
2. Baqui M, et al.: Human type 3 iodothyronine selenodeiodinase is located in the plasma membrane and undergoes rapid internalization to endosomes. J Biol Chem 2003; 278: 1206-11.
3. Baqui MM, et al.: Distinct subcellular localization of transiently expressed types 1 and 2 iodothyronine deiodinases as determined by immunofluorescence confocal microscopy. Endocrinology 2000; 141: 4309-12.
4. Curcio-Morelli C, et al.: In vivo dimerization of types 1, 2, and 3 iodothyronine selenodeiodinases. Endocrinology 2003; 144: 937-46.
5. Simpson GI, Leonard DM, Leonard JL: Identification of the key residues responsible for the assembly of selenodeiodinases. J Biol Chem 2006; 281: 14615-21.
6. Berry MJ, Banu L, Larsen PR: Type I iodothyronine deiodinase is a selenocysteine-containing enzyme. Nature 1991; 349: 438-40.
7. Croteau W, et al.: Cloning of the mammalian type II iodothyronine deiodinase. A selenoprotein differentially expressed and regulated in human and rat brain and other tissues. J Clin Invest 1996; 98: 405-17.
8. St Germain DL, et al.: A thyroid hormone-regulated gene in Xenopus laevis encodes a type III iodothyronine 5-deiodinase. Proc Natl Acad Sci U S A 1994; 91: 7767-71.
9. Bianco AC, et al.: Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev 2002; 23: 38-89.
10. Visser TJ, et al.: Subcellular localization of a rat liver enzyme converting thyroxine into tri-iodothyronine and possible involvement of essential thiol groups. Biochem J 1976; 157: 479-82.
11. Hesch RD, Brunner G, Soling HD: Conversion of thyroxine (T4) and triiodothyronine (T3) and the subcellular localisation of the converting enzyme. Clin Chim Acta 1975; 59: 209-213.
12. Mandel SJ, et al.: Cloning and in vitro expression of the human selenoprotein, type I iodothyronine deiodinase. J Clin Endocrinol Metab 1992; 75: 1133-9.
13. Köhrle J: Local activation and inactivation of thyroid hormones: the deiodinase family. Mol Cell Endocrinol 1999; 151: 103-19.
14. Koenig RJ: Regulation of type 1 Iodothyronine deiodinase in health and disease. Thyroid 2005; 15: 835-40.
15. Leonard JL, et al.: Localization of type I iodothyronine 5´-deiodinase to the basolateral plasma membrane in renal cortical epithelial cells. J Biol Chem 1991; 266: 11262-9.
16. Maia AL, et al.: Type 2 iodothyronine deiodinase is the major source of plasma T3 in euthyroid humans. J Clin Invest 2005; 115: 2524-33.
17. Jakobs TC, et al.: Structure of the human type I iodothyronine 5´-deiodinase gene and localization to chromosome 1p32-p33. Genomics 1997; 42: 361-3.
18. Toyoda N, et al.: A novel retinoid X receptor-independent thyroid hormone response element is present in the human type 1 deiodinase gene. Mol Cell Biol 1995; 15: 5100-12.
19. Jakobs TC, et al.: The promoter of the human type I 5´-deiodinase gene-mapping of the transcription start site and identification of a DR+4 thyroid-hormone-responsive element. Eur J Biochem 1997; 247: 288-97.
20. Zhang CY, et al.: Further characterization of thyroid hormone response elements in the human type 1 iodothyronine deiodinase gene. Endocrinology 1998; 139: 1156-63.
21. de Souza Meyer EL, et al.: Decreased type 1 iodothyronine deiodinase expression might be an early and discrete event in thyroid cell dedifferentation towards papillary carcinoma. Clin Endocrinol (Oxf) 2005; 62: 672-8.
22. Ambroziak M, et al.: Disturbed expression of type 1 and type 2 iodothyronine deiodinase as well as titf1/nkx2-1 and pax-8 transcription factor genes in papillary thyroid cancer. Thyroid 2005; 15: 1137-46.
23. Arnaldi LA, et al.: Gene expression profiles reveal that DCN, DIO1, and DIO2 are underexpressed in benign and malignant thyroid tumors. Thyroid 2005; 15: 210-21.
24. Huang Y, et al.: Gene expression in papillary thyroid carcinoma reveals highly consistent profiles. Proc Natl Acad Sci U S A 2001; 98(26): 15044-9.
25. Nauman A, et al.: Aktywność T4-5´-dejodynazy w raku jasnokomórkowym nerki. Urologia Polska Supp. 1991; 339-342.
26. Pachucki J, et al.: Type I 5´-iodothyronine deiodinase activity and mRNA are remarkably reduced in renal clear cell carcinoma. J Endocrinol Invest 2001; 24: 253-61.
27. Nauman P, et al.: The concentration of thyroid hormones and activities of iodothyronine deiodinases are altered in human brain gliomas. Folia Neuropathol 2004; 42: 67-73.
28. Debski MG, et al.: Human breast cancer tissue expresses high level of type 1 5´-deiodinase. Thyroid 2007; 17: 3-10.
29. Garcia-Solis P, Aceves C: 5´Deiodinase in two breast cancer cell lines: effect of triiodothyronine, isoproterenol and retinoids. Mol Cell Endocrinol 2003; 201: 25-31.
30. Nauman A, i wsp.: Aktywność T4-5´-dejodynazy w raku jasnokomórkowym nerki. Urologia Polska Supp. 1991; 339-342
31. Pietrzak M, et al.: 5´-deiodinase type I gene (hdioI) is abnormally activated by thyroid hormone receptor mutants cloned from human kidney cancers. European Journal of Biochemistry 2003; Supplement, abstract number 1.2 P30.
32. Piekielko-Witkowska A, et al.: The expression of alternatively spliced forms of type 1 deiodinase is changed in clear cell renal cell carcinoma. European Congress of Endocrinology 2007; Endocrine Abstracts 2007, 14 P119.
33. Puzianowska-Kuznicka M, et al.: Expression of thyroid hormone receptors is disturbed in human renal clear cell carcinoma. Cancer Lett 2000; 155: 145-52.
34. Kamiya Y, et al.: Expression of mutant thyroid hormone nuclear receptors is associated with human renal clear cell carcinoma. Carcinogenesis 2002; 23: 25-33.
35. Lin KH, et al.: Expression of mutant thyroid hormone nuclear receptors in human hepatocellular carcinoma cells. Mol Carcinog 1999; 26: 53-61.
36. Lin KH, Wu YH, Chen SL: Impaired interaction of mutant thyroid hormone receptors associated with human hepatocellular carcinoma with transcriptional coregulators. Endocrinology 2001; 142: 653-62.
37. Puzianowska-Kuznicka M, et al.: Functionally impaired thyroid hormone receptor mutants are present in thyroid papillary cancer. J Clin Endocrinol Metab 2002; 87: 1120-8.
38. Chan IH, Privalsky ML.: Thyroid hormone receptors mutated in liver cancer function as distorted antimorphs. Oncogene 2006; 25: 3576-88.
39. Frankton S, et al.: Multiple messenger ribonucleic acid variants regulate cell-specific expression of human thyroid hormone receptor beta1. Mol Endocrinol. 2004 18:1631-42.
40. Frankton S, et al.: Multiple messenger ribonucleic acid variants regulate cell-specific expression of human thyroid hormone receptor beta1. Mol Endocrinol. 2004;18(7):1631-42.
41. Hughes TA: Regulation of gene expression by alternative untranslated regions. Trends Genet. 2006;22(3):119-22.
42. Shyu AB, Wilkinson MF, van Hoof A: Messenger RNA regulation: to translate or to degrade. EMBO J. 2008;27(3):471-81.
43. Kozak M: How strong is the case for regulation of the initiation step of translation by elements at the 3´ end of eukaryotic mRNAs? Gene. 2004 Dec 8;343(1):41-54.
44. Filipowicz W, Bhattacharyya SN, Sonenberg N: Mechanisms of post-transcriptional regulation by microRNAs: are the answers insight? Nat Rev Genet. 2008;9(2):102-14.
45. Jaskiewicz L, Filipowicz W: Role of Dicer in posttranscriptional RNA silencing. Curr Top Microbiol Immunol. 2008; 320:77-97.
46. Morris KV. RNA-mediated transcriptional gene silencing in human cells. Curr Top Microbiol Immunol. 2008; 320:211-24.
47. Barbarotto E, Schmittgen TD, Calin GA: MicroRNAs and cancer: profile, profile, profile. Int J Cancer. 2008;122(5):969-77.
48. Cowland JB, Hother C, Grřnbaek K: MicroRNAs and cancer. APMIS. 2007 115(10):1090-106.
49. Skaftnesmo KO, et al.: MicroRNAs in tumorigenesis. Curr Pharm Biotechnol. 2007 8(6):320-5.
50. Master A, et al.: The expression of alternatively spliced 5´-UTR mRNA variants of human thyroid hormone receptor beta 1 (THRB1) is dependent on the tissue type and aberrant in clear cell renal cell carcinoma (ccRCC). Hormone Research, 2007; 48-49.
51. Bedrosian I, et al.: Deregulation alters the biologic properties of ovarian cancer cells. Oncogene 2004; 23: 2648-2657.
52. Nauman A, et al.: Elevated cyclin E level in human clear cell Renal Cell carcinoma: possible causes and consequences. Acta Biochimica Polonica 2007; 54: 595-602.
53. Bieda M, et al.: Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res 2006; 16: 595-605.
54. Pierce AM, et al.: E2F1 has both oncogenic and tumor-suppressive properties in a transgenic model. Mol Cell Biol 1999; 19: 6408-14.
55. DeGregori J, et al.: Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci USA 1997; 94: 7245-7250.
56. Hallstrom TC, Nevins JR: Specificity in the activation and control of transcription factor E2F-dependent apoptosis. Proc Natl Acad Sci USA 2003; 100: 10848-53.
57. Sherr CJ. Cancer cell cycles. Science 1996; 274: 1672-7.
58. Geng Y, et al.: Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene 1996; 12: 1173-80.
59. Helin K: Regulation of cell proliferation by the E2F transcription factors. Curr Opin Genet Dev 1998; 8: 28-35.
60. Moller MB, et al.: Frequent disruption of the RB1 pathway in diffuse large B cell lymphoma: prognostic significance of E2F-1 and p16INK4A. Leukemia 2000; 14: 898-904.
61. Turowska O, et al.: Overexpression of E2F1 in Clear Cell Renal Cell Carcinoma: A Potential Impact of Erroneous Regulation by Thyroid Hormone Nuclear Receptors. Thyroid 2007; 17: 1039-48.
62. Poplawski P, Nauman A: triiodothyronine leads to increased proliferation of clear cell renal cell carcinoma cells by changing expression of retinoblastoma family proteins EMBO Workshop 2007 „Molecular Mechanisms of cell cycle control in normal and malignant cells”, abstrakt nr 63.
63. Nicol D, et al.: Vascular endothelial growth factor expression is increased in renal cell carcinoma. J Urol 1997; 157(4): 1482-6.
64. Djordjevic G, et al.: Prognostic significance of vascular endothelial growth factor expression in clear cell renal cell carcinoma. Pathol Res Pract 2007; 203(2): 99-106.
65. Gnarra JR, et al.: Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 1994; 7: 85-90.
66. van den Berg A, et al.: Analysis of multiple renal cell adenomas and carcinomas suggests allelic loss at 3p21 to be a prerequisite for malignant development. Genes Chromosomes Cancer 1997; 19: 228-32.
67. Pietrzykowski A: Receptory jądrowe dla trijodotyroniny (TRβα1 i TRβ1) i dla kwasu retinowego (RARβ) w raku jasnokomórkowym nerki ludzkiej. Rozprawa doktorska (1997), (Biblioteka CMKP).
68. Nauman A, et al.: The low T3 syndrom in patients with kidney cancer – effect of cancer differentiation. Polish J Endocrinol 1996; 47: 365-74.
69. Tański Z: Zespół niskiej trijodotyroniny u chorych na raka nerki zależność od rozmiaru guza i stopnia jego zróżnicowania. Urologia Polska 2000; 53/3.
70. De Groot L: Dangerous dogmas in medicine: the nonthyroidal illness syndrome. J Clin Endocrinol Metab 1999, 84: 151-164.
otrzymano/received: 2008-03-25 zaakceptowano/accepted: 2008-05-10 Adres/address: *Alicja Nauman Zakład Biochemii i Biologii Molekularnej Centrum Medyczne Kształcenia Podyplomowego ul. Marymoncka 99/103, 01-813 Warszawa tel.: (0-22) 569-38-13 e-mail: anauman@cmkp.edu.pl The whole paper Zaburzenia szlaków sygnałowych hormonu tarczycy – trijodotyroniny – w raku nerki typu jasnokomórkowego is also available at On-line Medical Library. |
  Please click on selected cover
 |






Chcesz być na bieżąco? Polub nas na Facebooku: strona Wydawnictwa na Facebooku |