| Publisher |

|
published since 1994 |
| Publisher |
|
published since 1994 |
» Czynniki kontaktu w fizjologicznym krzepnięciu krwi oraz w zakrzepicy**© Borgis - Nowa Medycyna 2, p. 72-79
*Michał B. Ponczek, Michał Bijak
Czynniki kontaktu w fizjologicznym krzepnięciu krwi oraz w zakrzepicy**
Contact factors in blood clot formation and in thrombosis
Katedra Biochemii Ogólnej, Wydział Biologii i Ochrony Środowiska, Uniwersytet Łódzki
Kierownik Katedry: dr hab. Paweł Nowak Summary
A unique mechanism of clot formation evolved in time of the emergence of vertebrates, probably more than 500 million years ago. In the classical cascade of blood clotting thrombin formation may be initiated by two overlapping coagulation pathways – extrinsic and intrinsic pathway. The common target for these alternative routes is the initiation of coagulation factors X and IX, and next, in both cases, the activation of thrombin, which converts soluble fibrinogen into insoluble fibrin. Extrinsic pathway appears to be evolutionarily older, because the basic elements such as factor VII, tissue factor, and duplicated ancestor, descendant of factor IX and X, along with thrombin and fibrinogen are already appearing in primitive vertebrates such as jawless lampreys. Intrinsic pathway, also known as activation by contact, is evolutionarily much younger and is formed only in the ancestor of placental mammals and marsupials. Activation of blood coagulation by the contact is an invention of mammalian evolution, and in the remaining groups of terrestrial vertebrates plasma kinin-kallikrein system has no connection with the hemostatic system. New experimental and clinical observations confirm the protective role of contact factor deficiencies in certain types of disorders associated with thromboembolic disorders. The increased activity of the contact system is accompanied by life-threatening states such as sepsis, congenital angioedema or allergic conditions. A new approach to prevent and treat thromboembolic disease should take into account the contact factors, especially factor XI, but also factor XII and plasma prekallikrein, as novel targets for antithrombotic therapy, giving the opportunity to develop methods minimizing the bleeding complications. Key words: contact factors, factor XI, factor XII, prekallikrein
WSTĘP
Unikatowy mechanizm formowania skrzepu z fibrynogenu pod wpływem trombiny wyewoluował wraz z pojawieniem się kręgowców prawdopodobnie ponad 500 milionów lat temu (1). Ta nowa cecha umożliwiła sprawne zatrzymywanie wycieku krwi po uszkodzeniu powłoki ciała, przy równoczesnym zachowaniu ciągłości przepływu krwi w samych naczyniach układu krążenia (2). System ten, wymagany do zachowania równowagi pomiędzy zestalaniem krwi a jej płynnością, jest określany jako hemostaza. Zakłócenie tej równowagi prowadzi do zakrzepic lub niekontrolowanych krwawień. Aktywacja krzepnięcia krwi wiedzie poprzez kolejne przekształcanie zymogenów do aktywnych proteaz serynowych, z których kluczowe jest powstanie z protrombiny trombiny. Reakcje aktywacji zymogenów do proteaz wymagają specyficznych kofaktorów białkowych, fosfolipidów i jonów wapnia. Jedną z najważniejszych funkcji trombiny jest przekształcanie fibrynogenu w fibrynę oraz aktywacja płytek krwi, składników zarówno skrzepów, jak i skrzeplin (3).
W klasycznej kaskadzie krzepnięcia krwi tworzenie trombiny może zostać zapoczątkowane przez dwie zazębiające się drogi aktywacji krzepnięcia – szlak zewnątrzpochodny i wewnątrzpochodny (4-6). Wspólnym celem dla tych alternatywnych dróg inicjacji krzepnięcia są osoczowe czynniki X i IX, po czym dalej dochodzi, w obu przypadkach, do aktywacji trombiny. Oba czynniki wykazują identyczną masę cząsteczkową (tab. 1), budowę domenową, 44% podobieństwo sekwencji aminokwasowej oraz mogą być bezpośrednio aktywowane przez inicjator szlaku zewnątrzpochodnego – aktywny kompleks czynnika VII i tkankowego (7, 8), co wskazuje, że powstały prawdopodobnie na skutek duplikacji.
Tabela 1. Charakterystyka zymogenów proteaz kaskady krzepnięcia krwi człowieka.
Szlak zewnątrzpochodny wydaje się ewolucyjnie starszy, gdyż podstawowe elementy takie jak czynnik VII, czynnik tkankowy oraz zduplikowany potomek przodka czynnika IX i X, wraz z trombiną i fibrynogenem pojawiają się już u prymitywnych bezżuchwowych kręgowców takich jak minóg (9). Szlak wewnątrzpochodny z funkcjonalnym czynnikiem XI jest ewolucyjnie dużo młodszy i formuje się dodatkowo dopiero u przodka ssaków łożyskowych i torbaczy (10).
Procesem przeciwnym do kaskady krzepnięcia krwi jest fibrynoliza, zapoczątkowywana niemalże równocześnie z inicjacją krzepnięcia. Służy zapobieżeniu niepotrzebnemu i groźnemu w skutkach powstawaniu skrzeplin w obrębie układu krwionośnego oraz rozpuszczaniu czopu fibryny i płytek krwi tam, gdzie jest on już niepotrzebny. Proces ten zależny jest od plazminogenu oraz dwóch aktywatorów (tkankowego i urokinazowego aktywatora plazminogenu, tPA i uPA) przekształcających wymieniony zymogen do plazminy rozpuszczającej fibrynę (11). Fibrynoliza jest silnie powiązana z krzepnięciem nie tylko na etapie aktywacji plazminogenu przez tPA na fibrynie jako finalnym produkcie krzepnięcia (12), ale już na samym początku aktywacji krzepnięcia, w czym prawdopodobnie pewną rolę odgrywają czynniki wewnątrzpochodnego szlaku krzepnięcia krwi (13, 14). Dodatkowo, w obrębie kaskady krzepnięcia krwi działają inhibitory czynników krzepnięcia, takie jak inhibitor szlaku zależnego od czynnika tkankowego (TFPI), antytrombina III czy białka C i S. Jeśli mimo to wewnątrz naczynia krwionośnego żywego organizmu dojdzie do aktywacji krzepnięcia krwi, mamy do czynienia z zakrzepicą, którą określa się często jako „hemostaza w złym miejscu” (15).
Niezależnie od tego jak złożone są procesy zakrzepowe prowadzące do niedrożności naczyń krwionośnych, zwiększone generowanie trombiny, jako odpowiedź na czynnik prokoagulacyjny, lub osłabienie mechanizmów regulacyjnych uznaje się w wielu przypadkach za ważny element zakrzepic (16). Zgodnie z tym uzasadnieniem większość docelowych leków przeciwzakrzepowych skierowanych jest przeciwko trombinie lub też przeciwko innym zależnym od witaminy K proteazom serynowym (np. takim jak przede wszystkim czynnik X), które są wymagane do jej generowania. Zatem większość obecnie stosowanych leków skupia się na zewnątrzpochodnym szlaku aktywacji krwi, w którym poczynając od dostępnego po uszkodzeniu naczynia eksponowanego na tkankach czynnika tkankowego, sygnał przekazywany jest za pośrednictwem aktywowania zależnych od witaminy K zymogenów proteaz serynowych – czynnika VII, X i IX do protrombiny (16). Wynika to zapewne z tego, że ten właśnie szlak jest od lat uważany za główny mechanizm aktywacji krwi w hemostazie. Choć w praktyce klinicznej stosowanie tego typu terapii jest zazwyczaj skuteczne w zapobieganiu lub ograniczaniu wzrostu skrzepliny, zbyt często pojawia się poważne ryzyko nadmiernego krwawienia. Najnowsze badania myszy z wyłączonymi czynnikami krzepnięcia IX, XI i XII wykazały, że zakrzepica nie jest idealnym odbiciem lustrzanym prawidłowego krzepnięcia towarzyszącego zranieniom (17-21).
Także dane kliniczne dotyczące hemostazy i chorób zakrzepowo-zatorowych u ludzi potwierdzają tezę, że tor wewnątrzpochodny krzepnięcia krwi, w którym sygnał przekazywany jest od czynnika XII, poprzez czynnik XI, IX i X do protrombiny, może odgrywać ważną rolę w zakrzepicach (22), a czynniki kontaktu mogłyby zostać wykorzystane jako kolejny cel leków nowej generacji (23).
WEWNĄTRZPOCHODNY I ZEWNĄTRZPOCHODNY SZLAK KRZEPNIĘCIA KRWI A TWORZENIE FIBRYNY
Na wewnątrzpochodny tor krzepnięcia krwi składają się białka określane jako „czynniki kontaktu”, do których zalicza się osoczowe czynniki XI, XII, prekalikreinę osoczową i wielkocząsteczkowy kininogen. Łączą one układ hemostazy z osoczowym układem kininogenezy. O ile czynnik XII i prekalikreina osoczowa występują już u płazów i gadów (ptaki straciły czynnik XII prawdopodobnie w wyniku utraty genu), czynnik XI, białko kluczowe do zespolenia z układem krzepnięcia, pojawia się dopiero u ssaków, co można stwierdzić na podstawie analizy genomów przedstawicieli odpowiednich taksonów kręgowców (10). Aktywacja krzepnięcia krwi przez kontakt jest zatem wynalazkiem ewolucyjnym ssaków, a u pozostałych grup kręgowców lądowych osoczowy układ kininogenezy nie ma łączności z układem hemostazy (24, 25). Brak aktywacji krzepnięcia krwi z udziałem wewnątrzpochodnego szlaku u ptaków i gadów potwierdzają także wyniki eksperymentalne (25-28).
Nazwa „czynniki kontaktu” wywodzi się od tego, że czynnik XII ulega autoaktywacji „na powierzchni” w procesie nazywanym aktywacją krzepnięcia przez kontakt. Jako powierzchnię odpowiedzialną za aktywację uważało się in vivo pierwotnie kolagen, a in vitro funkcję taką spełnia kaolin (29). Obecnie jako alternatywne „fizjologiczne powierzchnie” rozważa się ujemnie naładowane polianiony, reszty fosforanowe uwalnianych z uszkodzonych komórek cząsteczek RNA (30) oraz nieprawidłowo pofałdowane białka, z tym że te ostatnie nie mają prowadzić do aktywacji krzepnięcia, lecz jedynie do uruchamiania układu kininogenezy (31). Natura fizyczna „kontaktu” wciąż nie jest do końca wyjaśniona, a nowe badania wskazują na pewne nieścisłości w standardowym paradygmacie ujemnie naładowanej powierzchni. Okazuje się, że autoaktywacja czynnika XII na powierzchni kontaktu nie jest w rzeczywistości specyficzna dla anionowych powierzchni hydrofilowych, a reakcja jest moderowana raczej przez skład białek fazy roztworu, w którym zachodzi (32).
Aktywacja czynnika XII do XIIa prowadzi przez aktywację czynnika XI do aktywacji czynnika IX, a ten z kolei działa na czynnik X. Kolejne aktywacje czynników XI, IX i X zachodzą przy udziale fosfolipidów płytek krwi (kompleksy związane z błoną płytkową). Czynnik XIIa przekształca także prekalikreinę osoczową do kalikreiny, a ta przekształca kolejne porcje czynnika XII do XIIa oraz trawi wielkocząsteczkowy kininogen do kinin. Sam wielkocząsteczkowy kininogen, poza prekursorem kinin, jest kofaktorem czynnika XII i prekalikreiny w aktywacji przez kontakt (32). Uwalnianie kinin przez układ kininogenezy jest niezależnym mechanizmem powodującym rozkurcz naczyń po ich uszkodzeniu, co prowadzi do zmniejszenia szybkości przepływu krwi w obszarze zranienia. Ułatwia to tworzenia czopu hemostatycznego poprzez agregację płytek krwi i polimeryzację fibryny (33).
Zapoczątkowanie procesów prowadzących do tworzenia fibryny w ramach zewnątrzpochodnego szlaku krzepnięcia krwi następuje po uszkodzeniu śródbłonka, gdy w osoczu czynnik VII tworzy kompleks z czynnikiem tkankowym, integralnym białkiem błony komórkowej. Czynnika tkankowego nie znajduje się zwykle w dużych stężeniach we krwi, ale jest on obecny w błonach komórkowych w podśródbłonkowej warstwie naczynia krwionośnego. Szlaki wewnątrz- i zewnątrzpochodny zbiegają się na poziomie aktywacji czynnika X, odpowiednio przez aktywny czynnik IX w obecności kofaktora czynnika VIIIa lub przez aktywny kompleks czynnika VII. Czynnik Xa aktywuje protrombinę do trombiny w obecności kofaktora czynnika Va, a następnie ta przekształca fibrynogen do fibryny (4) (ryc. 1).
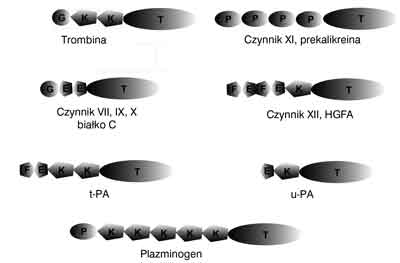 Ryc. 1. Budowa domenowa proteaz serynowych układu hemostazy. Domeny: T – domeny trypsynowe (proteazy serynowej); P – domeny „jabłkowe” (PAN); K – domeny kringlowe; F – domeny fibronektynowe; E – domeny czynnika wzrostu naskórka (EGF).
BUDOWA CZYNNIKÓW KONTAKTU
Wszystkie czynniki kontaktu są glikoproteinami i za wyjątkiem wielkocząsteczkowego kininogenu należą do proteaz serynowych, podobnie jak większość białek głównego, zewnątrzpochodnego szlaku krzepnięcia. Proteazy kontaktu wyróżnia jednakże fragment N-końcowy występujący przed domeną proteazową. Dwa z nich – czynnik XI oraz prekalikreina osoczowa – dzielą ze sobą identyczny fragment złożony z 4 domen PAN (ryc. 2) (nazywanych także domenami „jabłkowymi” – ang. apple domains) oraz znaczne, aż 60% podobieństwo całej sekwencji aminokwasowej. Fakt ten, wraz z lokalizacją genów dla czynnika XI oraz prekalikreiny osoczowej tuż obok siebie na tym samym chromosomie oraz brakiem czynnika XI u kręgowców innych niż ssaki, świadczy o niedawnej duplikacji lokalnej, która doprowadziła do rozdzielenia się opisywanych białek u przodka ssaków (10). Czynnik XI odróżnia jednakże od prekalikreiny znacząco ważna dla jego aktywności cecha strukturalna. Czynnik XI formuje homodimer tworzony poprzez mostki disulfidowe cystein czwartej domeny PAN dwóch różnych łańcuchów polipeptydowych (Cys321 wg numeracji białka człowieka) (34). Prekalikreina osoczowa posiada dodatkową resztę cysteiny (w pozycji 326 wg numeracji białka człowieka), tworząc z Cys321 wewnątrzłańcuchowe wiązanie disulfidowe (co tym samym uniemożliwia tworzenie homodimerów) (35). Cecha ta jest także charakterystyczna dla przodków PK/FXI u kręgowców niebędących ssakami (10). Mutacja Cys326 u przodka czynnika XI prawdopodobnie w glicynę nadała temu białku wyjątkową możliwość formowania homodimerów, co jest niezwykle istotne dla jego aktywacji (34, 36, 37).
 Ryc. 2. Schematyczne przedstawienie układu hemostazy człowieka (opracowanie oryginalne).
F – czynniki krzepnięcia krwi, FBG – fibrynogen, FBN – fibryna, FBX – fibryna stabilizowana przez czynnik XIII, FDP – produkty degradacji fibryny, FDPX – produkty degradacji fibryny stabilizowanej, KNG – wielkocząsteczkowy kininogen, PK – prekalikreina osoczowa, PL – fosfolipidy, PLG – plazminogen, PLN – plazmina, t-PA – tkankowy aktywator plazminogenu, u-PA – urokinazowy aktywator plazminogenu. Procesy związane z krzepnięciem – linie ciągłe, procesy związane z fibrynolizą – linie przerywane. W odróżnieniu od wymienionych białek czynnik XII na N-końcu posiada 2 domeny fibronektynowe, 2 naskórkowego czynnika wzrostu i 1 kringlową (ryc. 2). Pojawia się już u płazów i wykazuje podobieństwo procentowe i strukturalne do aktywatora czynnika wzrostu hepatocytów (HGFA), z którym dzieli prawdopodobnie pochodzenie od wspólnego czworonożnego przodka kręgowców lądowych (38). Geny dla obu białek zlokalizowane są na odrębnych chromosomach, co świadczy o duplikacji chromosomu (10).
Od wymienionych dotychczas białek kontaktu odróżnia się wielkocząsteczkowy kininogen, należący do kininogenów, a nie proteaz. W pełni funkcjonalny kininogen, z segmentem bogatym w reszty histydyny, pojawia się dopiero u płazów, co zgadza się z pojawieniem się czynnika XII w tej właśnie gromadzie kręgowców (39).
ROLA CZYNNIKÓW KONTAKTU W HEMOSTAZIE I ZAKRZEPICY
W obecnych modelach hemostazy uważa się, że tworzenie fibryny w miejscu zranienia wywołane jest przede wszystkim przez kompleks czynnik tkankowy–czynnik VIIa i dalej czynnik X, a czynnik XII nie jest kategorycznie wymagany, choć uznaje się rolę czynników XI i IX we wzmocnieniu i podtrzymaniu krzepnięcia (40-43). Mimo powszechnego uznania szlaku zewnątrzpochodnego jako przeważającego w krzepnięciu krwi in vivo, niektóre obserwacje kliniczne są sprzeczne z tak uproszczonym modelem. Objawy krwotoczne u osób cierpiących na niedobory czynnika IX (hemofilia B) i jego kofaktora czynnika VIII (hemofilia A) wskazują na występowanie znacznie bardziej skomplikowanych interakcji obejmujących oba szlaki krzepnięcia. Po pierwsze, w przeciwieństwie do czynników IX i VIII, niedobór czynnika XI (hemofilia C) wiąże się zwykle ze znacznie łagodniejszymi zaburzeniami krzepnięcia, charakteryzującymi się niewielkimi krwotokami okołooperacyjnymi, przede wszystkim w obrębie tkanek o wysokiej aktywności układu fibrynolitycznego oraz zwykle brakiem spontanicznych krwotoków (44, 45). Po drugie, pacjenci z niedoborem czynnika XII nie wykazują tendencji do nieprawidłowego krwawienia, nawet w trakcie operacji chirurgicznych, mimo znacznie dłuższego czasu kaolinowo-kefalinowego (APTT) (46). Wymiennie obserwacje przeczą poprawności modelu, w którym proteazy krzepnięcia aktywowane są wyłącznie w sposób liniowy.
Niedobory czynników zewnątrzpochodnego szlaku krzepnięcia, takich jak czynnik VII i czynnik tkankowy, wiążą się z poważnymi krwotokami, wskazując na niewątpliwe kluczowe znaczenie tego szlaku w prawidłowym krzepnięciu krwi (47, 48). Jednakże aktywność kompleksów czynnika VII z czynnikiem tkankowym jest ściśle regulowana przez TFPI (inhibitor drogi zależnej od czynnika tkankowego), który inaktywuje kompleks VIIa/TF na błonach komórkowych. Dlatego do prawidłowego formowania włóknika potrzebny jest u ssaków także alternatywny szlak aktywacji trombiny z udziałem białek szlaku wewnątrzpochodnego. Jak już wcześniej wspomniano, wrodzony niedobór czynnika XI u człowieka skutkuje łagodnymi krwotokami, zazwyczaj w obrębie tkanek miękkich o wysokiej aktywności fibrynolitycznej, natomiast niedoborowi czynnika XII zwykle w ogóle nie towarzyszą zwiększone krwawienia. Wyjaśnieniem tego paradoksu jest obecność niezależnego mechanizmu aktywacji czynnika XI bezpośrednio przez trombinę, do czego czynnik XII jest zbędny (49, 50). Dzięki temu powstaje pętla sprzężenia zwrotnego prowadząca do zwiększonego generowania trombiny poprzez czynnik XI, co staje się niezależne od aktywacji wewnątrzpochodnej przez kontakt i także poza kontrolą szlaku zewnątrzpochodnego (ominięcie inhibitora TFPI). Odmienności w zaburzeniach krwawienia przy niedoborach czynników IX, XI i XII wskazują także, jak ważną rolę w prawidłowym krzepnięciu odgrywa czynnik IX, który może być aktywowany bezpośrednio przez kompleks VIIa/TF oraz przez czynnik XI aktywowany przez trombinę niezależnie od czynnika XII (ryc. 1) (51). Znaczenie czynników XI i IX w hemostazie i zakrzepicy potwierdzają badania prowadzone na szczepach myszy ze znokautowanymi genami odpowiednio dla czynnika IX i XI w modelu zakrzepicy tętnicy szyjnej wywołanej FeCl3. Chronione przed sztucznie generowanym zatorem w porównaniu ze szczepami dzikimi były nie tylko myszy ze znokautowanym czynnikiem IX, ale także te ze znokautowanym czynnikiem XI (51). W innych badaniach okazało się, że znokautowanie czynnika XI chroni także myszy przed generowanymi FeCl3 zakrzepami żyły głównej (45, 52). Równocześnie tylko w przypadku braku czynnika IX obserwowano wydłużenie czasu krwawienia ogona. Wskazywać to ma na potencjalne znaczenie czynnika XI jako celu działania nowych leczniczych inhibitorów zapobiegających zakrzepom, bez ingerencji w prawidłowe krzepnięcie krwi (23, 53).
Chociaż dane sugerują, że niedobór czynnika XI ma działanie ochronne przed zakrzepami, trudno ocenić wagę tej obserwacji w przypadku zakrzepicy u ludzi. FeCl3 wywołuje uszkodzenie warstwy śródbłonkowej naczynia, co prawdopodobnie prowadzi do znacznej ekspozycji kolagenu w stosunku do przepływającej krwi i ma niewielkie znaczenie dla rozpoczęcia zakrzepicy żylnej w dużych naczyniach. Nie jest też jasne, czy aktywowany czynnik XI działa, zwiększając po prostu całkowitą aktywność czynnika IX w miejscu uszkodzenia, czy działa, generując IXa w miejscach odziaływania zakrzep–naczynie. Aktywność in vitro czynnika XI jest istotna do zachowania integralności skrzepu w czasie, gdy osocze lub krew ulega krzepnięciu pod wpływem TF lub trombiny w obecności aktywatorów fibrynolizy. Ten efekt zachodzi przynajmniej częściowo za pośrednictwem generowania aktywnego czynnika XI za pośrednictwem trombiny, prowadząc do aktywacji metaloproteazy, inhibitora fibrynolizy TAFI (ang. thrombin-activable fibrinolysis inhibitor) (inna nazwa CPB2, karboksypeptydaza B2) (53).
Ochronne działanie zarówno przed formowaniem złogów włóknika, jak i agregacją płytek krwi w przypadku braku czynnika XI u myszy wykazano także, wywołując uszkodzenia naczyń za pomocą lasera i mikroskopii przyżyciowej (54). W tym przypadku okazało się, że efekt ochronny w największym stopniu dotyczył zmniejszenia agregacji płytek krwi w miejscu uszkodzenia, bo aż o 90% w porównaniu z 50% zmniejszeniem tworzenia złogów fibryny. Mimo że u znokautowanych myszy w miejscu uszkodzenia formowała się bogatopłytkowa skrzeplina, była jednak niestabilna i szybko rozpadała się, prowadząc do przywrócenia drożności naczynia. Przeciwzakrzepowy efekt braku czynnika XI nie wydaje się związany ze zmniejszeniem aktywacji TAFI, ponieważ myszy ze znokautowanym TAFI nie były chronione przed zatorami tętniczymi po uszkodzeniu wywołanym FeCl3 (37). Wykazano także, że u królików zahamowanie czynnika XI przez specyficzne przeciwciała zwiększało rozpuszczanie skrzeplin indukowane t-PA w modelu zakrzepicy żył ucha i w tętnicach biodrowych (55, 56). Także u naczelnych wykazano wagę czynnika XI w zakrzepicy, gdyż zablokowanie przeciwciałami chroniło przed wzrostem bogatopłytkowej skrzepliny w modelu pomostów tętniczo-żylnych u pawianów (52, 57). Powyższe badania wskazują, że czynnik XI wykazuje prozakrzepowe działanie u wielu gatunków ssaków.
Co ciekawe, także niedobór prekalikreiny osoczowej oraz czynnika XII u myszy chroni przed zakrzepicą żylną i tętniczą, nie wywołując negatywnych skutków w prawidłowym krzepnięciu, w przypadku czynnika XII zarówno w modelach udaru mózgu, jak i zawału serca (17, 58-61). Wskazywałoby to, że przynajmniej u myszy, w czasie uszkodzenia naczynia z towarzyszącym niedotlenieniem i reperfuzją (jak w modelu z FeCl3), czynnik XI jest aktywowany przez czynnik XII, prowadząc do wzrostu skrzepliny zgodnie z klasycznym wewnątrzpochodnym modelem krzepnięcia.
W przypadku człowieka podejrzewano, że niedobory czynnika XII, nie wywołując co prawda skaz krwotocznych, mogły być odpowiedzialne za wzrost ryzyka zakrzepic, co tłumaczono rolą tego białka w katalizowaniu konwersji plazminogenu do plazminy (13). Potwierdzać to miały obserwacje kliniczne, wraz z klasycznym przypadkiem Johna Hagemana, u którego po raz pierwszy wykryto niedobór czynnika XII i który jednak zmarł z powodu zatorowości płucnej po 12 dniach hospitalizacji od złamania kości miednicy (62, 63). Późniejsze badania nie potwierdziły jednak związku między niedoborem czynnika XII a zakrzepicami u człowieka (64-67).
Wymienione obserwacje kliniczne i eksperymentalne dotyczące czynników XI i XII sugerowałyby, że nie jest całkowicie poprawna koncepcja, w której formowanie skrzepliny odpowiada zaburzeniu równowagi normalnego procesu hemostazy działającego w miejscu zranienia (68). Przy wielu wspólnych elementach fizjologiczna hemostaza i patologiczna zakrzepica wydają się angażować nieco inne mechanizmy (59). Zewnątrzpochodny szlak krzepnięcia jest ważny zarówno dla hemostazy, jak i zakrzepicy, a niedobory lub zwiększona aktywność tego szlaku mogą prowadzić do krwotoków i zakrzepic (69).
Stabilizacja skrzeplin powodujących w systemie naczyniowym zatory wymaga dodatkowej aktywacji fibrynogenu oraz płytek krwi. Proces ten jest zależny od czynników XI i XII i ma mniejsze znaczenie w zapobieganiu krwawienia w miejscu zranienia. Pojawia się zatem możliwość terapeutycznego zastosowania inhibitorów wymienionych czynników w celu zapobiegania i leczenia zakrzepic, które nie wywoływałyby powikłań krwotocznych, co niestety obserwuje się podczas stosowania obecnych antykoagulantów (18). W przypadku człowieka takie podejście mogłoby być skuteczne w przypadku udarów niedokrwiennych, gdzie stwierdzono zmniejszoną zapadalność osób cierpiących na niedobór czynnika XI, w przeciwieństwie do przypadków zawału serca, gdzie takiej zależności nie wykazano (70, 71).
Dodatkowe ciekawe wnioski odnośnie wagi czynnika XI w formowaniu fibryny wynikają z badań myszy, u których nokautowi poddano dodatkowo geny elementów układu hemostazy istotnych dla prawidłowej fibrynolizy – białka C i plazminogenu. Myszy ze znokautowanymi genami odpowiednio dla białka C i plazminogenu bardzo wcześnie wykazują śmiertelne koagulopatie związane z tworzeniem złogów. Myszy bez białka C z dodatkowo znokautowanym czynnikiem XI żyją dłużej, lecz w przypadku braku plazminogenu przeżywają krócej. Przeżywalność myszy z niedoborem plazminogenu ulega poprawie przez nokaut czynnika IX. Potwierdza to dodatkowo, że czynnik XI jest bardzo ważny do tworzenia fibryny in vivo, ale zarazem może mieć dodatkowe funkcje w regulacji stanu zapalnego lub przy naprawie tkanek (72, 73).
Myszy z niedoborem czynnika XI zostały także zbadane w modelu sepsy bakteryjnej wywołanej przez podwiązanie i przebicie jelita ślepego (57). Niedobór czynnika XI zwiększał przeżywalność i redukował infiltrację leukocytów oraz koagulopatie, sugerując, że białko to przyczynia się do rozwoju stanu zapalnego, a jego farmakologiczne inhibitory mogą być korzystne w leczeniu sepsy (20, 21).
Wymienione właściwości czynnika XI czynią go ciekawym celem nowych leków zakrzepowych (74, 77, 78). Dodatkowo, brak krwotocznych skutków niedoboru czynnika XII zarówno u człowieka, jak i myszy, przy zanotowanej przeciwzakrzepowej aktywności niedoboru czynnika XII u myszy i braku potwierdzenia prozakrzepowego działania niedoboru czynnika XII u człowieka, wskazują na to białko jako na kolejny potencjalny cel leków przeciwzakrzepowych (23, 61).
PODSUMOWANIE
Od dawna znany i badany in vitro system kontaktu, nieco zaniedbany w badaniach in vivo i w praktyce klinicznej na rzecz zewnątrzpochodnego systemu krzepnięcia krwi, dzięki nowym badaniom na myszach powoli odsłania swoją tajemniczą rolę w patologicznym krzepnięciu krwi znanym pod zbiorczą nazwą „zakrzepic”. System ten rozpoczyna prozakrzepowe i prozapalne reakcje poprzez aktywację wewnątrzpochodną krzepnięcia krwi i kininogenezę, gdzie czynnikiem aktywującym może być nie tylko kolagen, ale także nieprawidłowo pofałdowane białka czy polifosforany, między innymi RNA (75). Nowe obserwacje eksperymentalne i kliniczne potwierdzają ochronną rolę niedoborów czynników kontaktu w pewnych typach zaburzeń zatorowych związanych z zakrzepicami. Zwiększona aktywność systemu kontaktu towarzyszy z kolei zagrażającym życiu zaburzeniom takim jak sepsa, wrodzony obrzęk naczynioruchowy czy stany alergiczne (58, 75, 76). Nowe podejście do profilaktyki i leczenia chorób zatorowo-zakrzepowych powinno uwzględnić czynniki kontaktu, przede wszystkim czynnik XI, ale także czynnik XII i prekalikreinę osoczową, jako nowe cele terapii przeciwzakrzepowych, dając możliwość opracowania metod minimalizujących powikłania krwotoczne (23)1.
**Praca ta nie powstałaby bez grantu stażowego EMBO ASTF-83-2008 oraz finansów na badania statutowe Uniwersytetu Łódzkiego 506/810. Pragniemy także podziękować profesorom Russellowi Doolittle i Barbarze Wachowicz za opiekę naukową.
1W październiku 2010 roku rozpoczęły się badania kliniczne preparatu ISIS-FXIRx w USA. Jest to potencjalny lek antysensowny skierowany na mRNA czynnika XI, przeznaczony do leczenia zakrzepic żylnych poprzez hamowanie syntezy czynnika XI w wątrobie (Identifier: NCT01713361, http://clinicaltrials.gov). Druga faza badań klinicznych rozpoczęła się w 2012 roku, a planowany termin zakończenia badań to luty 2014 roku. Piśmiennictwo
1. Shu D: Cambrian explosion: Birth of tree of animals. Gondwana Res 2008; 14: 219-240. 2. Davidson CJ, Tuddenham EG, McVey JH: 450 million years of hemostasis. J Thromb Haemost 2003; 1: 1487-1494. 3. Schenone M, Furie BC, Furie B: The blood coagulation cascade. Curr Opin Hematol 2004; 11: 272-277. 4. Riddel JPJ, Aouizerat BE, Miaskowski C et al.: Theories of blood coagulation. J Pediatr Oncol Nurs 2007; 24: 123-131. 5. Davie EW, Ratnoff OD: Waterfall sequence for intrinsic blood clotting clotting. Science 1964; 145: 1310-1312. 6. Furie B, Furie BC: The molecular-basis of blood-coagulation. Cell 1988; 53: 505-518. 7. Bauer KA, Kass BL, ten Cate H et al.: Factor IX is activated in vivo by the tissue factor mechanism. Blood 1990; 76: 731-736. 8. Komiyama Y, Pedersen AH, Kisiel W: Proteolytic activation of human factors IX and X by recombinant human factor VIIa: effects of calcium, phospholipids, and tissue factor. Biochemistry 1990; 29: 9418-9425. 9. Doolittle RF, Jiang Y, Nand J: Genomic evidence for a simpler clotting scheme in jawless vertebrates. J Mol Evol 2008; 66: 185-196. 10. Ponczek MB, Gailani D, Doolittle RF: Evolution of the contact phase of vertebrate blood coagulation. J Thromb Haemost 2008; 6: 1876-1883. 11. Cilia La Corte AL, Philippou H, Ariens RAS: Role of fibrin structure in thrombosis and vascular disease. Adv Protein Chem Str 2011; 83: 75-127. 12. Schielen WJ, Voskuilen M, Tesser GI et al.: The sequence A alpha-(148-160) in fibrin, but not in fibrinogen, is accessible to monoclonal antibodies. Proc Natl Acad Sci USA 1989; 86: 8951-8954. 13. Goldsmith GHJ, Saito H, Ratnoff OS: The activation of plasminogen by Hageman factor (Factor XII) and Hageman factor fragments. J Clin Invest 1978; 62: 54-60. 14. Laake K, Venneröd AM: Factor XII-induced fibrinolysis: studies on the separation of prekallikrein, plasminogen proactivator, and factor XI in human plasma. Thromb Res 1974; 4: 285-302. 15. Colman RW: Are hemostasis and thrombosis two sides of the same coin? J Exp Med 2006; 203: 493-495. 16. Brenner B, Hoffman R: Emerging options in the treatment of deep vein thrombosis and pulmonary embolism. Blood Rev 2011; 25: 215-221. 17. Renné T, Pozgajová M, Grüner S et al.: Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med 2005; 202: 271-281. 18. Renné T, Gailani D: Role of Factor XII in hemostasis and thrombosis: clinical implications. Expert Rev Cardiovasc Ther 2007; 5: 733-741. 19. Cheng QF, Tucker EI, Pine MS et al.: A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood 2010; 116: 3981-3989. 20. Tucker EI, Helm M, McCarty OJT et al.: Factor XI Inhibitor Antibody Treatment Improves Survival In a Murine Polymicrobial Sepsis Model. Blood 2010; 116: 4762-4768. 21. Gailani D, Tucker EI, Cheng Q et al.: Factor XI deficiency confers a survival advantage in a murine sepsis model. Blood 2006; 108: 299A-300A. 22. Blat Y, Seiffert D: A renaissance for the contact system in blood coagulation? Thromb Haemost 2008; 99: 457-460. 23. Muller F, Gailani D, Renne T: Factor XI and XII as antithrombotic targets. Curr Opin Hematol 2011; 18: 349-355. 24. Ponczek MB, Gailani D, Doolittle RF: Evolution of the contact phase of vertebrate blood coagulation. J Thromb Haemost 2008; 6: 1876-1883. 25. Conlon JM: Bradykinin and its receptors in non-mammalian vertebrates. Regul Pept 1999; 79: 71-81. 26. Soslau G, Wallace B, Vicente C et al.: Comparison of functional aspects of the coagulation cascade in human and sea turtle plasmas. Comp Biochem Physiol B Biochem Mol Biol 2004; 138: 399-406. 27. Weir-M J, Acurero Z, Salas-A R et al.: Blood coagulation factors in the black headed vulture (Coragyps atratus), a potential animal model for the study of haemostasis. Thromb Res 2004; 113: 269-273. 28. Frost CL, Naudé RJ, Oelofsen W et al.: Comparative blood coagulation studies in the ostrich. Immunopharmacology 1999; 45: 75-81. 29. van der Meijden PE, Munnix IC, Auger JM et al.: Dual role of collagen in factor XII – dependent thrombus formation. Blood 2009; 114: 881-90. Epub 2009/04/18. 30. Kannemeier C, Shibamiya A, Nakazawa F et al.: Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci USA 2007; 104: 6388-6393. 31. Maas C, Govers-Riemslag JWP, Bouma B et al.: Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest 2008; 118: 3208-3218. 32. Vogler EA, Siedlecki CA: Contact activation of blood-plasma coagulation. Biomaterials 2009; 30: 1857-1869. 33. Schmaier AH, McCrae KR: The plasma kallikrein-kinin system: its evolution from contact activation. J Thromb Haemost 2007; 5: 2323-2329. 34. Meijers JC, Mulvihill ER, Davie EW et al.: Apple four in human blood coagulation factor XI mediates dimer formation. Biochemistry 1992; 31: 4680-4684. 35. McMullen BA, Fujikawa K, Davie EW: Location of the disulfide bonds in human plasma prekallikrein: the presence of four novel apple domains in the amino-terminal portion of the molecule. Biochemistry 1991; 30: 2050-2056. 36. Wu W, Sinha D, Shikov S et al.: Factor XI homodimer structure is essential for normal proteolytic activation by factor XIIa, thrombin, and factor XIa. J Biol Chem 2008; 283: 18655-18664. 37. Gailani D, Smith SB: Structural and functional features of factor XI. J Thromb Haemost 2009; 7: 75-78. 38. Ponczek M: Czynniki kontaktu układu hemostazy: badanie ewolucji molekularnej oraz nowe perspektywy terapeutyczne. Postepy Biochem 2010; 56: 67-74. 39. Zhou L, Li-Ling J, Huang H et al.: Phylogenetic analysis of vertebrate kininogen genes. Genomics 2008; 91: 129-141. 40. Broze GJJ: Tissue factor pathway inhibitor and the current concept of blood coagulation. Blood Coagul Fibrinolysis 1995; 6 (suppl. 1): 7-13. 41. Mackman N: Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol 2004; 24: 1015-1022. 42. Rapaport SI, Rao LV: The tissue factor pathway: how it has become a “prima ballerina”. Thromb Haemost 1995; 74: 7-17. 43. Morrissey JH: Tissue factor: A key molecule in hemostatic and nonhemostatic systems. Int J Hematol 2004; 79: 103-108. 44. Cawthern KM, van’t Veer C, Lock JB et al.: Blood coagulation in hemophilia A and hemophilia C. Blood 1998; 91: 4581-4592. 45. Gomez K, Bolton-Maggs P: Factor XI deficiency. Haemophilia 2008; 14: 1183-1189. 46. Halbmayer WM, Haushofer A, Schon R et al.: The prevalence of moderate and severe fxii (hageman-factor) deficiency among the normal population – evaluation of the incidence of fxii deficiency among 300 healthy blood-donors. Thromb Haemost 1994; 71: 68-72. 47. Rosen ED, Chan JCY, Idusogie E et al.: Mice lacking factor VII develop normally but suffer fatal perinatal bleeding. Nature 1997; 390: 290-294. 48. Bugge TH, Xiao Q, Kombrinck KW et al.: Fatal embryonic bleeding events in mice lacking tissue factor, the cell-associated initiator of blood coagulation. Proc Natl Acad Sci USA 1996; 93: 6258-6263. 49. Matafonov A, Sarilla S, Sun MF et al.: Activation of factor XI by products of prothrombin activation. Blood 2011; 118: 437-445. 50. Wielders SJH, Beguin S, Hemker HC et al.: Factor XI – dependent reciprocal thrombin generation consolidates blood coagulation when tissue factor is not available. Arterioscler Thromb Vasc Biol 2004; 24: 1138-1142. 51. Wang X, Cheng Q, Xu L et al.: Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost 2005; 3: 695-702. 52. Tucker EI, Marzec UM, White TC et al.: Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood 2009; 113: 936-944. Epub 2008/10/24. 53. Renne T, Oschatz C, Seifert S et al.: Factor XI deficiency in animal models. J Thromb Haemost 2009; 7 (suppl. 1): 79-83. Epub 2009/07/28. 54. Furie B, Furie BC: In vivo thrombus formation. J Thromb Haemost 2007; 5 (suppl. 1): 12-17. Epub 2007/08/01. 55. Yamashita A, Nishihira K, Kitazawa T et al.: Factor XI contributes to thrombus propagation on injured neointima of the rabbit iliac artery. J Thromb Haemost 2006; 4: 1496-1501. Epub 2006/07/15. 56. Minnema MC, Friederich PW, Levi M et al.: Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI. In vivo evidence for a role of factor XI as an anti-fibrinolytic factor. J Clin Invest 1998; 101: 10-14. Epub 1998/02/14. 57. Tucker EI, Gailani D, Hurst S et al.: Survival advantage of coagulation factor XI-deficient mice during peritoneal sepsis. J Infect Dis 2008; 198: 271-274. 58. Revenko AS, Gao D, Crosby JR et al.: Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood 2011; 118: 5302-5311. 59. Kleinschnitz C, Stoll G, Bendszus M et al.: Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med 2006; 203: 513-518. 60. Kleinschnitz C, Stoll G, Bendszus M et al.: Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med 2006; 203: 513-8. 61. Renne T, Nieswandt B, Gailani D: The intrinsic pathway of coagulation is essential for thrombus stability in mice. Blood Cells Mol Dis 2006; 36: 148-151. 62. Ratnoff OD, Busse RJ, Sheon RP: The demise of John Hageman. N Engl J Med 1968; 279: 760-761. 63. Lammle B, Wuillemin WA, Huber I et al.: Thromboembolism and bleeding tendency in congenital factor XII deficiency – a study on 74 subjects from 14 Swiss families. Thromb Haemost 1991; 65: 117-121. 64. Girolami A, Randi ML, Gavasso S et al.: The occasional venous thromboses seen in patients with severe (homozygous) FXII deficiency are probably due to associated risk factors: a study of prevalence in 21 patients and review of the literature. J Thromb Thrombolysis 2004; 17: 139-143. 65. Girolami A, Morello M, Girolami B et al.: Myocardial infarction and arterial thrombosis in severe (homozygous) FXII deficiency: no apparent causative relation. Clin Appl Thromb Hemost 2005; 11: 49-53. 66. Zeerleder S, Schloesser M, Redondo M et al.: Reevaluation of the incidence of thromboembolic complications in congenital factor XII deficiency – a study on 73 subjects from 14 Swiss families. Thromb Haemost 1999; 82: 1240-1246. 67. Girolami A, Ruzzon E, Lombardi AM et al.: Thrombosis-free surgical procedures in severe (Homozygote) factor XII deficiency: report of four additional cases and literature review. Clin Appl Thromb Hemost 2004; 10: 351-355. 68. Gailani D, Renne T: Intrinsic pathway of coagulation and arterial thrombosis. Arterioscler Thromb Vasc Biol 2007; 27: 2507-2513. 69. Mackman N: Triggers, targets and treatments for thrombosis. Nature 2008; 451: 914-918. 70. Salomon O, Steinberg DM, Koren-Morag N et al.: Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood 2008; 111: 4113-4117. 71. Salomon O, Steinberg DM, Dardik R, et al.: Inherited factor XI deficiency confers no protection against acute myocardial infarction. J Thromb Haemost. 2003; 1: 658-661. 72. Cheng QF, Zhao Y, Lawson WE et al.: The effects of intrinsic pathway protease deficiencies on plasminogen-deficient mice. Blood 2005; 106: 3055-3057. 73. Chan JCY, Ganopolsky JG, Cornelissen I et al.: The characterization of mice with a targeted combined deficiency of protein C and factor XI. Am J Pathol 2001; 158: 469-479. 74. Karuturi R, Al-Horani RA, Mehta SC et al.: Discovery of allosteric modulators of factor XIa by targeting hydrophobic domains adjacent to its heparin-binding site. J Med Chem 2013; 56: 2415-2428. 75. Maas C, Oschatz C, Renne T: The Plasma Contact System 2.0. Semin Thromb Hemost 2011; 37: 375-381. 76. Renné T: The procoagulant and proinflammatory plasma contact system. Semin Immunopathol 2012; 34: 31-41. 77. Zhang H, Löwenberg EC, Crosby JR et al.: Inhibition of the intrinsic coagulation pathway factor XI by antisense oligonucleotides: a novel antithrombotic strategy with lowered bleeding risk. Blood. 2010; 116: 4684-4692. 78. Younis HS, Crosby J, Huh JI et al.: Antisense inhibition of coagulation factor XI prolongs APTT without increased bleeding risk in cynomolgus monkeys. Blood 2012; 119: 2401-2408.
otrzymano/received: 2012-12-12 zaakceptowano/accepted: 2013-02-25 Adres/address: *Michał B. Ponczek Katedra Biochemii Ogólnej Wydział Biologii i Ochrony Środowiska Uniwersytet Łódzki ul. Pomorska 141/143, 90-236 Łódź tel.: +48 (42) 635-44-82, fax: +48 (42) 635-44-84 e-mail: mponczek@biol.uni.lodz.pl, mbijak@biol.uni.lodz.pl Paper Contact factors in blood clot formation and in thrombosis at On-line Medical Library. |
|||||||||||||||||||||||||||||||||||||||||||

 wersja polska
wersja polska









