© Borgis - Medycyna Rodzinna 3, p. 11-15
*Sergiusz Jóźwiak1, Monika Dudzisz-Śledź2
Stwardnienie guzowate – możliwości diagnostyczne i terapeutyczne
Tuberous sclerosis complex – diagnostic and therapeutic possibilities
1Klinika Neurologii i Epileptologii Instytutu „Pomnik Zdrowia Dziecka” w Warszawie
Kierownik Kliniki: prof. dr hab. n. med. Sergiusz Jóźwiak
2Novartis Poland Sp. z o.o., Warszawa
Kierownik: dyrektor medyczny dr n. med. Jan Krzysztof Żelazowski
Summary
Tuberous sclerosis complex (TSC) is one of the most common neurocutaneous diseases. The prevalence of TSC is 1:6000 in general population. The disease belongs to neuroectodermal dysplasias known as phacomatoses. TSC is the result of genes TSC1 and TSC2 mutations. The disease is characterized by diverse clinical phenotype and prolonged and progressive course. The most common clinical features of this disease include epilepsy and developmental delay. It should be emphasized, that mental development is normal in almost 50% patients. Genetically determined potential risk of developing tuberous lesions, hamartomas or benign tumors of different organs: heart, kidney, brain, liver, skin is characteristic for this condition. Signs and symptoms appear in different periods of development, some of them could appear in the prenatal life. The diagnosis of TSC is based on precisely defined clinical criteria and imaging studies. The patients should be closely controlled with imaging studies due to increased risk of tumors development. The treatment of TSC is primarily symptomatic, comprehensive and based on involved organs. It should be conducted in specialized centers. Especially problematic is epilepsy management. Epilepsy is often drug-resistant and needs to be treated with combined therapy. The treatment should be conducted under supervision of experienced neurologist. Recently big progress has been made regarding TSC diagnosis, monitoring and treatment, leading to significant improvement of care of patients with TSC.
Key words: tuberous sclerosis complex, epilepsy, mental retardation, cardiac tumors
Wstęp
Stwardnienie guzowate (TSC, ang. tuberous sclerosis complex) in. choroba Bourneville'a należy do grupy chorób określanych wspólnym mianem fakomatozy lub dysplazje neuroektodermalne, które charakteryzują się występowaniem zaburzeń rozwojowych w obrębie trzech listków zarodkowych, a zwłaszcza ektodermy. Częstość występowania choroby szacowana jest na 1: 6 000 (1). Na całym świecie na tę chorobę choruje 1,5 miliona ludzi (2). Choroba odznacza się zróżnicowanym obrazem klinicznym, a do jej charakterystycznych objawów należą: padaczka, opóźnienie rozwoju, zmiany skórne oraz zmiany rozrostowe w układzie nerwowym, narządzie wzroku, nerkach, sercu i wątrobie. Choroba ma charakter przewlekły i postępujący (1, 3).
Choroba jest dziedziczona jako cecha autosomalna dominująca z wysoką penetracją i zmienną ekspresją genu, co jest powodem dużej różnorodności obrazu klinicznego. Za jej rozwój odpowiedzialne są mutacje dwóch genów: TSC1 (9q34) i TSC2 (16p13), przy czym mutacje genu TSC2 związane są z cięższym przebiegiem choroby (1, 4, 5, 6). Mutacje w obrębie genów TSC1 i TSC2 stwierdzane są u około 85% pacjentów spełniających kliniczne kryteria rozpoznania TSC. U około 15% pacjentów z TSC nie stwierdza się jednak żadnej mutacji w obrębie genów TSC1 i TSC2, a rozpoznanie stawiane jest na podstawie obrazu klinicznego. Przyczyną tego faktu może być mozaikowatość gonadalna (zmutowane komórki są tylko w obrębie gonad i u pozornie zdrowej osoby nie stwierdza się cech stwardnienia guzowatego) lub też udział innych mechanizmów w patogenezie TSC. Nie wiadomo również, dlaczego te same mutacje u różnych osób warunkują odmienny obraz i przebieg kliniczny choroby, a więc osoby o tym samym genotypie mogą mieć różne cechy fenotypowe (7, 8). W około 60% (9) – 70% (8) przypadków stwierdza się nowe mutacje. Objawy chorobowe pojawiają się wraz ze wzrostem dziecka, a u dzieci w pierwszym roku życia rozpoznanie choroby może być szczególnie trudne (8). Geny TSC1 i TSC2 należą do genów supresorowych nowotworów, czyli hamujących rozwój nowotworów. Białka kodowane przez te geny, hamartyna i tuberyna, odgrywają istotną rolę w hamowaniu szlaku przekazywania sygnałów przebiegającego z udziałem kinazy mTOR (ang. mammalian target of rapamycin) (8). Aby doszło do wystąpienia choroby, konieczna jest inaktywacja obu prawidłowych alleli genu TSC1 lub TSC2 (10).
W ciągu ostatnich dwudziestu lat dokonał się olbrzymi postęp w zakresie diagnostyki, monitorowania przebiegu i leczenia stwardnienia guzowatego, co doprowadziło do istotnej poprawy opieki nad pacjentami z TSC (4, 7). Na podstawie licznych badań przedklinicznych ustalono, iż hamartyna i tuberyna pełnią funkcje regulatorowe szlaków przekazywania sygnałów Rheb i mTOR. Odkrycia te pozwoliły na rozpoczęcie dalszych prac nad molekularnymi mechanizmami choroby oraz nad możliwościami ich leczenia z uwzględnieniem inhibitorów kinazy mTOR (ang. mammalian target of rapamycin) (7, 11).
Stwardnienie guzowate jest wyjątkowo skomplikowaną chorobą, dotykającą różnych układów narządów w różnym stopniu i w zróżnicowany sposób, w różnym okresie życia pacjentów (4).
Obraz kliniczny
Objawy kliniczne, które wskazują na rozpoznanie TSC są bardzo różnorodne i w dużej mierze uzależnione są od wieku pacjenta. U dzieci objawami, które naprowadzają lekarza na to rozpoznanie są najczęściej guzki ( rhabdomyosarcoma) serca lub drgawki, u dorosłych są to zwykle zmiany skórne, nerkowe lub płucne. Często choroba rozpoznawana jest dopiero u pacjenta w wieku dorosłym, po wystąpieniu drgawek lub krwotoku z guza nerki (4).
Najczęstsze zmiany w TSC dotyczą OUN i skóry. Mózg i skóra zajęte są u 90-95% pacjentów z TSC (4, 7). Większość zmian w przebiegu TSC uwidacznia się u dzieci powyżej 3. roku życia, co utrudnia ustalenie rozpoznania we wcześniejszym wieku (7).
Do głównych zmian skórnych zalicza się: znamiona bezbarwne (ryc. 1), mnogie guzkowe naczyniakowłókniaki na twarzy (ryc. 2), płaskie włókniaki w okolicy czołowej, zmiany na skórze określane jako „skóra szagrynowa”, czyli mnogie zmiany o średnicy 0,5-10 cm zwykle barwy cielistobrązowej przypominające włókniaki (ryc. 3), rozsiane na tułowiu szczególnie w okolicy lędźwiowo-krzyżowej, włókniaki okołopaznokciowe, uszypułowane włókniaki okolicy karkowej i szyjnej, zmiany typu „confetti” czyli liczne plamiste drobne odbarwienia o średnicy 1-3 mm zwykle w okolicy przedramion i podudzi (12, 13).
 Ryc. 1. Plama odbarwieniowa na skórze w przebiegu TSC.
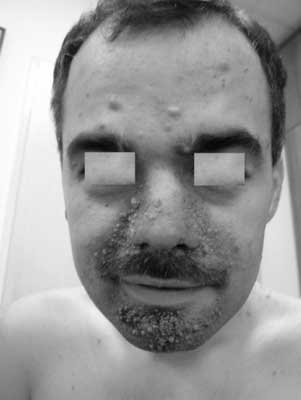 Ryc. 2. Naczyniakowłókniaki na twarzy u mężczyzny z TSC. Ryc. 2. Naczyniakowłókniaki na twarzy u mężczyzny z TSC.
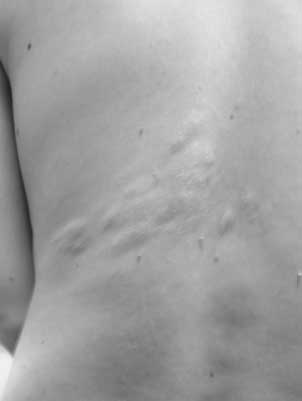 Ryc. 3. Skóra szagrynowa u pacjenta z TSC. Ryc. 3. Skóra szagrynowa u pacjenta z TSC.
Do zmian narządowych w przebiegu TSC należą torbiele i nowotwory łagodne o wolnym tempie wzrastania rozwijające się w obrębie nerek, serca, mózgu i narządu wzroku (12).
Ze strony OUN są to wapniejące guzki na brzegach komór bocznych mózgu oraz guzki korowo-podkorowe mózgu widoczne jedynie w MRI mózgu (ryc. 4). Rosnące podwyściółkowe guzki okołokomorowe mogą niekiedy prowadzić do rozwoju wodogłowia (9). U 90% pacjentów z TSC występuje padaczka, która u 70% pacjentów pojawia się w pierwszym roku życia. Pacjenci z podejrzeniem TSC powinni być regularnie obserwowani pod kątem padaczki, zwłaszcza w pierwszym roku życia. Padaczka jest często oporna na leczenie, a opóźnienie skutecznego leczenia prowadzi do nieodwracalnych zmian w OUN, których konsekwencją są zaburzenia rozwoju dziecka. Ponadto występują zaburzenia poznawcze (u 50% pacjentów), zaburzenia z kręgu autyzmu, zaburzenia snu, problemy psychiczne oraz zaburzenia zachowania i problemy wychowawcze, spowodowane m.in. zaburzeniami koncentracji, artykulacji i myślenia abstrakcyjnego (4, 7, 14).
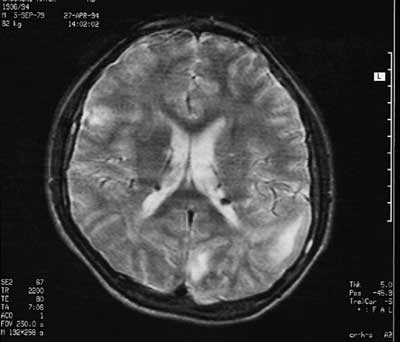 Ryc. 4. Guzki okołokomorowe i guzki korowe widoczne w obrazie MRI pacjenta z TSC. Ryc. 4. Guzki okołokomorowe i guzki korowe widoczne w obrazie MRI pacjenta z TSC.
Zmiany w nerkach występują u około 70-90% pacjentów (7). W ponad połowie przypadków zmiany w nerkach stwierdzane są przypadkowo podczas okresowej kontroli pacjentów, kiedy to stwierdza się zmianę echogeniczności nerek w badaniu ultrasonograficznym bądź obniżenie klirensu kreatyniny endogennej (14). Najczęściej stwierdzaną zmianą w nerkach są naczyniakomięśniakotłuszczaki ( angiomyolipoma). Guzy te mogą występować, jako samodzielna zmiana chorobowa, nie związana z TSC, lub też w przebiegu TSC (14). Ponadto występują torbiele nerek, rak jasnokomórkowy nerki, oncocytoma, torbiele okołonerkowe oraz wielotorbielowatość nerek (13). Zmiany w nerkach mogą prowadzić do zmniejszenia ilości czynnego miąższu nerkowego. Są one obserwowane u trzech czwartych pacjentów z TSC z równą częstością wśród płci męskiej i żeńskiej. Angiomyolipoma nerek w przebiegu TSC u ok. 60% pacjentów są bezobjawowe. Mogą być one przyczyną dolegliwości bólowych oraz źródłem krwawienia (13). Częstość występowania guzów nerek w TSC wzrasta z wiekiem, a prawdopodobieństwo występowania objawów klinicznych związanych z tymi guzami wzrasta w przypadku wielkości angiomyolipoma powyżej 4 cm (9).
Guzy rhabdomyoma serca to najczęstsze guzy serca stwierdzane u dzieci, również w życiu płodowym. Guzy te często występują w przebiegu TSC, aczkolwiek częstość ich występowania jest różna w zależności od wieku, w jakim rozpoznano chorobę. Guzy te mają skłonność do regresji z upływem czasu, najszybszą regresję ich wielkości obserwuje się w ciągu pierwszych trzech lat życia dziecka. Mogą być one przyczyną zaburzeń rytmu serca (7, 15).
Zmiany ze strony płuc mogą, aczkolwiek nie muszą być pierwszą manifestacją choroby. Lymphangioleiomyomatoza (LAM) występuje u pacjentów z TSC rzadko, ale charakteryzuje się wysoką chorobowością i śmiertelnością (16). LAM występuje prawie wyłącznie u kobiet w wieku rozrodczym, u mężczyzn właściwie nie występuje. Najczęściej objawia się dusznością spowodowaną odmą opłucnową lub chylothoraks, przewlekłym kaszlem, krwiopluciem, bólem w klatce piersiowej i świstami. Znane są również przypadki bezobjawowe. LAM wyjątkowo rzadko występuje u pacjentów bez TSC. Polega na proliferacji mięśni gładkich przewodzików i oskrzelików oraz naczyń w obrębie płuc i prowadzi do niewydolności oddechowej. W badaniach czynnościowych płuc częściej stwierdza się zmiany obturacyjne aniżeli restrykcyjne (16). Średni czas przeżycia pacjentek z LAM wynosi około 10 lat (16).
Hamartoma siatkówki stwierdzane są u ok. połowy pacjentów z TSC (7, 13). W większości przypadków zmiany są łatwo stwierdzane podczas badania dna oka (13). Ich występowanie w przebiegu TSC nie wykazuje korelacji z wiekiem pacjenta (13).
Rozpoznanie
Kryteria rozpoznania stwardnienia guzowatego zostały przedstawione w tabeli 1. Rozpoznanie jest pewne, gdy spełnione są 2 duże kryteria lub 1 duże i 2 małe, a prawdopodobne, gdy spełnione jest 1 duże i 1 małe kryterium. Rozpoznanie jest możliwe przy występowaniu 1 dużego lub co najmniej dwóch małych objawów (8).
Tabela 1. Kryteria rozpoznawania stwardnienia guzowatego (5, 7, 8).
| Kryteria duże | Kryteria małe |
Naczyniakowłókniaki (angiofibroma) skóry twarzy lub płaski włókniak na czole
Atraumatyczne włókniaki okołopaznokciowe
Znamiona bezbarwne (3 lub więcej)
Ogniska skóry szagrynowej
Mnogie hamartoma siatkówki
Guzki korowe mózgu
Guzki podwyściółkowe mózgu
Podwyściółkowy gwiaździak olbrzymiokomórkowy
Guzy serca pojedyncze lub mnogie
Lymphangiomatosis płuc
Angiomyolipoma nerek | Mnogie ubytki szkliwa
Polipy odbytu
Torbiele kości
Ogniska migracji istoty białej mózgu
Włókniaki dziąseł
Hamartoma o lokalizacji pozanerkowej
Zmiany w siatkówce oka
Plamy na skórze typu "confetti"
Mnogie torbiele nerek |
W praktyce klinicznej do rozpoznania wystarczy stwierdzenie objawów klinicznych i odchyleń w badaniach dodatkowych. Metody diagnostyki genetycznej, chociaż dostępne, nie są wymagane do ustalenia rozpoznania i nie zostały ujęte w kryteriach diagnostycznych (4, 17).
Diagnostyka
Do badań diagnostycznych wykonywanych u pacjentów z podejrzeniem TSC należą: usg (a w szczególnych przypadkach CT lub MRI) jamy brzusznej, nerek, rtg płuc, ekg, echo serca, badanie okulistyczne w tym badanie dna oka, EEG, CT i MRI mózgu, rtg układu kostnego, badania psychologiczne oraz neuropsychologiczne (5, 7). Badania obrazowe odgrywają coraz większą rolę w diagnostyce, leczeniu i monitorowaniu przebiegu TSC (2, 7).
Badania genetyczne pozwalają ustalić typ mutacji będącej przyczyną TSC. Są one również pomocne w ocenie ryzyka wystąpienia najpoważniejszych powikłań u pacjentów z TSC. Pozwalają one na: (1) potwierdzenie rozpoznania, (2) ustalenie czy mutacja ma charakter spontaniczny czy też jest dziedziczona, (3) ustalenie rozpoznania w życiu płodowym (diagnostyka prenatalna) w przypadku rodzin, w których choroba już występuje (7).
Badanie MRI mózgu powinno być wykonane przed ukończeniem drugiego roku życia dziecka (7).
Badanie elektroencefalograficzne (EEG) odgrywa zasadniczą rolę w rozpoznawaniu padaczki u pacjentów z TSC (4). Jest ono szczególnie przydatne jeśli TSC zamanifestowało się w pierwszej kolejności pod postacią napadów drgawkowych. Dzieci i młodzież, u których nigdy nie występowały napady drgawkowe na ogół nie wymagają wykonywania tego badania (7). W przypadku dzieci poniżej 1. roku życia, u których TSC zostało zdiagnozowane przed wystąpieniem drgawek, szczególne znaczenie ma monitorowanie elektroencefalograficzne w celu wczesnego wykrycia ewentualnych zmian napadowych (7).
W diagnostyce rhabdomyoma serca zasadnicze znaczenie odgrywa echokardiografia, której wykonanie zalecane jest w przypadku wystąpienia objawów ze strony serca oraz w przypadku zaburzeń jego funkcji. Ponadto w momencie ustalenia rozpoznania TSC wskazane jest wykonanie badania elektrokardiograficznego (7).
W przypadku podejrzenia LAM najlepszą metodą diagnostyczną wydaje się być HRCT (tomografia komputerowa wysokiej rozdzielczości), która zwykle powinna być wykonana u dorosłych kobiet z TSC (7). Przydatne są również badania czynnościowe płuc, w których częściej stwierdza się zaburzenia typu obturacyjncego, a rzadziej typu restrykcyjnego (16).
Diagnostyka zmian nerkowych w momencie ustalania rozpoznania obejmuje badanie ultrasonograficzne oraz badania CT bądź MRI (2, 7).
Przy ustalaniu rozpoznania TSC wykonywane są również badania okulistyczne oraz badanie neuropsychologiczne (7).
Badania kontrolne
Po ustaleniu rozpoznania wskazane jest regularne wykonywanie badań kontrolnych. Badania neuroobrazowe powinny być wykonywane co roku do ukończenia 21. roku życia, jeśli u pacjenta występują kliniczne lub stwierdzane badaniami obrazowymi czynniki ryzyka wystąpienia gwiaździaka (7). W innych przypadkach wskazane jest wykonanie badań obrazujących OUN w odstępach co najmniej 1-3-letnich w celu wychwycenia niepokojącego wzrostu nowotworów. Wskazana jest regularna obserwacja pacjenta w dzieciństwie i w wieku dojrzewania pod kątem objawów klinicznych ze strony OUN oraz pod kątem pojawienia się szybko rosnącej zmiany w OUN. Może to pozwolić na interwencję we wczesnej fazie rozwoju gwiaździaków podwyściółkowych, które mogą prowadzić do powstawania wodogłowia (7, 18).
Badania ultrasonograficzne powinny być wykonywane co najmniej co 1-3 lata, aby uchwycić niepokojący wzrost nowotworów (7).
Badanie CT klatki piersiowej jest powtarzane w przypadku wystąpienia zaburzeń ze strony układu oddechowego (7).
Badanie elektrokardiograficzne powtarzane jest w razie istnienia wskazań klinicznych, natomiast badanie echokardiograficzne w przypadku zaburzeń funkcji serca (7).
Badanie EEG po ustaleniu rozpoznania wykonywane jest w razie pojawienia się wskazań klinicznych. (7).
Diagnostyka okulistyczna oraz neuropsychologiczna powtarzane są w razie potrzeby klinicznej. Badanie neuropsychologiczne powtarzane jest również, kiedy dziecko rozpoczyna edukację szkolną (7).
Leczenie
Leczenie pacjentów chorych na stwardnienie guzowate jest trudne, kompleksowe i jak dotychczas w głównej mierze objawowe (7). Obejmuje ono przede wszystkim usuwanie nowotworów, laseroterapię i leczenie chirurgiczne zmian skórnych, leczenie padaczki, wsparcie psychologiczne. Leczenie to powinno być prowadzone w ośrodkach specjalistycznych, zwłaszcza ze względu na złożony i trudny do przewidzenia obraz kliniczny choroby i związaną z tym konieczność regularnych kontroli za pomocą m.in. specjalistycznych badań obrazowych.
Leczenie padaczki stanowi istotne wyzwanie u pacjentów z TSC. Padaczka w przebiegu TSC powinna być leczona przez doświadczonego neurologa, a terapia powinna być rozpoczęta jak najwcześniej. Szczególnie duże znaczenie ma jak najwcześniejsze wykrycie padaczki, aby zapobiec następstwom nieleczonych napadów. W leczeniu stosowane są środki przeciwdrgawkowe, najczęściej wigabatryna. U wielu pacjentów padaczka jest oporna na leczenie farmakologiczne, nawet przy zastosowaniu terapii skojarzonej dwoma lub trzema preparatami. U trzech czwartych pacjentów z padaczką w przebiegu TSC dochodzi do wystąpienia padaczki opornej na leczenie. Oprócz metod farmakologicznych stosowane jest niefarmakologiczne leczenie padaczki, zwłaszcza opornej na leczenie. Obejmuje ono stosowanie diety ketogennej (wysokotłuszowa, ubogowęglowodanowa), stymulację nerwu błędnego za pomocą specjalnego urządzenia podobnego do stymulatora oraz leczenie chirurgiczne polegające na usunięciu zmiany będącej przyczyną napadów drgawkowych (4, 19).
Leczenie LAM jest bardzo trudne, zasadniczo objawowe i polega na stosowaniu m.in. beta-mimetyków rozszerzających oskrzela. Podejmowane są również próby terapii hormonalnej, a w chorobie zaawansowanej transplantacja płuc (16).
Duże nadzieje wiąże się obecnie ze stosowaniem inhibitorów szlaku mTOR (pochodnych rapamycyny) w hamowaniu wzrostu zmian narządowych. Wstępne publikacje na ten temat są bardzo obiecujące (7, 20). Piśmiennictwo
1. Jóźwiak J, Jóźwiak S, Włodarski P: Possible mechanisms of disease development in tuberous sclerosis. Lancet Oncol 2008; 9: 73-79. 2. Henry J, Baskin Jr: The pathogenesis and imaging of the tuberous sclerosis complex. Pediatr Radiol 2008; 38: 936-952. 3. Domańska-Pakieła D, Kotulska K, Jóźwiak S: Stwardnienie guzowate: postępy w diagnostyce i leczeniu. Neurologia dziecięca 2008; 17(33): 11- 22. 4. Kwiatkowski DJ, Whittemore VH, Thiele EA: Tuberous Sclerosis Complex: Genes, Clinical Features and Therapeutics. 2010, WILEY-VCH Verlag GmbH&Co. KGaA Weinheim. ISBN: 978-3-527-32201-5. 5. Miłońska S, Murak-Kozanecka E: Stwardnienie guzowate – opis przypadku. Postępy Psychiatrii i Neurologii 2008; 17(2): 161-164. 6. Niida Y et al.: Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests diff erent genetic mechanisms for pathogenesis of TSC lesions. Am J Hum Genet 2001; 69: 493-503. 7. Curatolo P, Bombardieri R, Jóźwiak S: Tuberous sclerosis. Lancet 2008; 372: 657-668. 8. Jóźwiak S: Opieka nad chorym ze stwardnieniem guzowatym – zmiany narządowe i zalecane badania kontrolne. Str. 92-100. Folium, Lublin 2004, wyd. 1, ISBN 83-87991-38-4. 9. Roach ES, Sparagana SP: Diagnosis of tuberous sclerosis complex. J Child Neurol 2004; 19: 643-649. 10. Crino P B et al.: Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology 2010; 74: 1716- 1723. 11. Kevin C: Ess. Tuberous sclerosis complex: everything old is new again. J Neurodevelop Disord 2009; 1: 141-149. 12. Jóźwiak S: Medycyna Praktyczna Pediatria 1999/06 Pediatria ilustrowana, odcinek 6: Zmiany skórne i narządowe w stwardnieniu guzowatym u dzieci. Str. 77. 13. Lendvay T, Marshall FF: The tuberous sclerosis complex and its highly variable manifestations. The Journal of Urology 2003; 169: 1635-1642. 14. Szczepańska M et al.: Powikłania nefrologiczne w zespole stwardnienia guzowatego. Wiadomości lekarskie 2007; LX, 9-10: 483-488. 15. Tworetzky W et al.: Association Between Cardiac Tumors and Tuberous Sclerosis in the Fetus and Neonate. Am J Cardiol 2003; 92: 487-489. 16. Hancoc E et al.: Lymphangioleiomyomatosis and tuberous sclerosis. Respiratory Medicine 2002; 96: 7-13. 17. Farfał S et al.: Stwardnienie guzowate – klinika, diagnostyka, leczenie. Pol Merk Lek 2004; 16: 589-591. 18. Anisya-Vasanth AV et al.: Spectrum of epilepsy in tuberous sclerosis. Neurology India 2004; 52(2): 210-212. 19. Bebin EM, Kelly PJ, Gomez MR: Surgical treatment for epilepsy in cerebral tuberous sclerosis. Epilepsia 1993; 34(4): 651-657. 20. Lam C et al.: Rapamycin (Sirolimus) in Tuberous Sclerosis Associated Pediatric Central Nervous System Tumors. Pediatr Blood Cancer 2010; 54: 476-479.
otrzymano/received: 2010-08-11
zaakceptowano/accepted: 2010-08-31
Adres/address:
*Sergiusz Jóźwiak
Klinika Neurologii i Epileptologii Instytutu "Pomnik Zdrowia Dziecka" w Warszawie
Al. Dzieci Polskich 20, 04-730 Warszawa
tel.: (22) 815 74 04
e-mail: sergiusz.jozwiak@gmail.com
|
|