© Borgis - Medycyna Rodzinna 2, p. 31-38
*Paweł Kowalczyk
Gen p53jako hamulec molekularny przeciwdziałający powstawaniu nowotworów
The p53gen as molecular brake a counteragent of tumor formation
Interdyscyplinarne Centrum Modelowania Matematycznego i Komputerowego Uniwersytetu Warszawskiego
Dyrektor Placówki: prof. dr hab. Marek Niezgódka
Summary
Neoplastic transformation in cancer depends on accumulation of alterations in many families of genes that control signal transduction, cell proliferation and genomic stability. These changes observed in nature generally involve two groups of genes with opposite functions: oncogenes and tumor supressor genes. There are many tumor suppressor genes that are inactivated in almost every type of human cancer, most important being TP53.
The p53protein is involved in the regulation of cell cycle, apoptosis and cell differentiation. p53plays a central role in a complex DNA damage-sensing network which, in response to genotoxic stress such as ionizing radiation, either shunts the cell into a prolonged G1 arrest, presumably to allow for DNA repair and/or directs the cell along the apoptotic pathway. The p53tumour suppressor gene has proven to be one of the genes most often mutated in human cancers (about 50%) what demonstrate their role in the development of cancers.
It involves mainly point mutations leading to amino acid substitutions in the central region of the protein which impair its normal functions. Analysis of the mutational events that target the p53gene has revealed evidence for both exogenous and endogenous mutational mechanisms. More than 90% of the mutations reported so far are clustered between exons 4 and 9. This region is highly conserved throughout evolution and contains the DNA binding domain of the protein which is essential to p53functional activity.
Key words: human p53gene, DNA damage, colorectal, breast and prostate cancer, trans-4-hydroxy-2-nonenal, lipid peroxidation
Powstawanie nowotworów
Materiał genetyczny komórek organizmów żywych jest stale narażony na uszkodzenia i modyfikacje, co może w konsekwencji prowadzić do mutacji. Te ostatnie mogą spontanicznie powstawać w wyniku błędów popełnionych podczas replikacji DNA. Są to jednak przypadki niezwykle rzadkie. Częstość mutacji wzrasta, gdy komórki są narażone na działanie mutagenów – czynników chemicznych (np. aldehyd chlorooctowy) lub fizycznych (np. promieniowanie jonizujące i ultrafioletowe), które reagując bezpośrednio z DNA mogą wywoływać różnorodne zmiany w materiale genetycznym, np. modyfikacje zasad, wiązania krzyżowe, przerwy w podwójnym łańcuchu. Również działanie reaktywnych form tlenu (RFT) i azotu (RFA), wytwarzanych podczas procesu zapalnego może powodować uszkodzenie wielu składników komórki w tym lipidów, prowadząc do ich peroksydacji – których głównym składnikiem jest trans -4-hydroxy-2-nonenal (HNE). Wszystkie te czynniki odgrywają istotną rolę w mutagenezie, kancerogenezie oraz w procesie starzenia – powodując rozwój nowotworu. Powstawanie nowotworu zaczyna się od mutacji w obrębie komórek normalnych. Mutacja zwiększa skłonność komórek do namnażania się, podczas gdy sąsiednie komórki pozostają w stanie spoczynku. Zmieniona komórka i jej komórki potomne nadal wyglądają prawidłowo, ale dzielą się nadmiernie – wykazując hiperplazję (nadmierny rozrost). W ciągu kilku lat, jedna na milion tych komórek ulega kolejnej mutacji (pod wpływem czynników endo- lub egzogennych), która może spowodować rozluźnienie kontroli wzrostu komórkowego. Oprócz nadmiernej proliferacji potomstwo tej komórki różni się pod względem kształtu i wyglądu od normalnych komórek, wykazując dysplazję (nieprawidłowy wygląd ograniczony do warstwy nabłonkowej), po pewnym czasie zachodzi kolejna rzadka mutacja, która zmienia zachowanie komórek. Komórki dotknięte mutacjami są coraz bardziej nieprawidłowe pod względem wzrostu i wyglądu, przekształcając się w komórki nowotworowe. Jeśli nowotwór nie naruszył jeszcze granic między tkankami mówimy że jest to rak in situ (rak przedinwazyjny). Nowotwór ten może pozostać w takim stanie bardzo długo, ale w niektórych komórkach czasami zachodzą dodatkowe mutacje. Gdy zmiany genetyczne umożliwiają nowotworowi rozpoczęcie inwazji na sąsiednie tkanki i rozsiewanie komórek do krwi lub limfy – nowotwór jest w pełni złośliwy. Komórki które wymknęły się spod kontroli, mogą doprowadzić do powstawania w organizmie nowych guzów (przerzutów), które mogą zaburzyć biologię komórek jeszcze nie zainfekowanych, prowadząc do ich transformacji nowotworowej, co w konsekwencji prowadzi do śmierci organizmu, wskutek zniszczenia tkanek w różnych narządach.
Wśród genów, których mutacja przyczynia się do rozwoju nowotworu można wyróżnić: protoonkogeny (1), geny supresorowe, zwane także antyonkogenami (2) oraz geny zwiększające częstość mutacji w genomie (3). Na szczególną uwagę zasługuje ludzki gen supresorowy określony jako p53. Mutacja obecna w tym genie jest najczęstszym defektem genetycznym stwierdzonym w ponad połowie wszystkich typów nowotworów. Transformacja nowotworowa wywołana przez defekt genu p53pojawia się w przypadku zadziałania dwustopniowego mechanizmu: inaktywacji białka p53na skutek mutacji punktowej w jednym allelu i utraty drugiego allelu. Produkt genu p53znany jest pod nazwami „białko p53” oraz „białko TP p53”.
Struktura genu p53
W haploidalnym genomie komórek organizmu człowieka, występuje pojedynczy gen p53, który zlokalizowano w chromosomie 17p, w strefach 12-13, 3. Mutacje w tym rejonie genu mogą powodować chorobę genetyczną nazwaną zespołem Li-Fraumeni (2). Gen p53składa się z 11 egzonów o łącznej masie 20 kpz. Strukturę pierwszorzędową białka p53poznano sekwencjonując cDNA (4). Masa cząsteczkowa białka p53człowieka wynosi 53 000 Da (stąd wyprowadzono nazwę genu i jego produktu). Natywne białko p53jest fosfoproteiną o składającą się z 393 aminokwasów zorganizowanych w trzy domeny (ryc. 1). N-końcowa domena (bogata w aminokwasy kwaśne) aktywuje transkrypcję, środkowa (rdzeniowa) wiąże się do specyficznych sekwencji DNA, natomiast C-końcowa (bogata w aminokwasy zasadowe) jest odpowiedzialna na tetrameryzacje białka umożliwiającą wiązanie się do promotorów, które zawierają wiele powtórzeń sekwencji palindromowej złożonej z 10 par zasad. Ta cecha powoduje zwykle aktywację (rzadziej inaktywację) transkrypcji DNA na mRNA. W wiązaniu domeny rdzeniowej p53do DNA kluczową rolę odgrywa sześć aminokwasów: 5 reszt argininy oraz reszta glicyny. Dwie argininy bezpośrednio oddziałują z helisą DNA, a pozostałe pełnią rolę stabilizującą. Aminokwasy te są częstym celem mutagenów, gdyż ich „wyłączenie” uniemożliwia transaktywację genów przez p53.
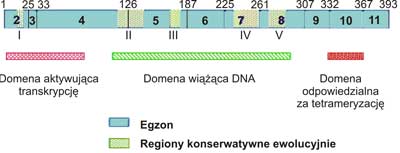
Ryc. 1. Struktura ludzkiego genu p53.
W komórkach myszy również znaleziono gen p53w chromosomie 11. Gen ten składa się z 11 eksonów i zawiera on około 12,5 kpz. W chromosomie 14 zidentyfikowano ponadto bezintronowy pseudogen p53, niezdolny do produkcji pełnej długości białka. Organizacja genu p53 człowieka i myszy jest podobna. Jednakże ludzki gen p53jest większy od funkcjonalnego genu myszy zawiera bowiem 20,3 kpz (Lamb P. 1986). Porównanie sekwencji ludzkiego i mysiego cDNA białka wskazuje na 78% homologię. Największą homologię (92%) stwierdza się w obrębie kodonów 156-288. Przypuszcza się, że ten właśnie region jest szczególnie ważny dla funkcji tego białka. Wykryto polimorficzne formy białka p53człowieka, które wynikały z różnic w strukturze kodonów 72 i 21 (6).
Mutacje w genie p53
W ponad połowie przypadków nowotworów u ludzi stwierdzono mutacje w genie p53 (3). Rozrzut mutacji badany wzdłuż całego genu nie jest równomierny. Mutacje występują głównie w obrębie eksonów 5-8, gdzie znajdują się cztery domeny o wysokiej konserwatywności (HCD II-V). Wyróżnia się 3, 4 lub nawet 5 do 6 gorących miejsc ( hot spot region, HSR), czyli regionów podatnych na mutacje (7) (ryc. 2). W wyszczególnionych pięciu regionach (gorących miejscach) wykrywa się ponad 70% wszystkich mutacji w genie p53. Dla porównania, w genie ras z rodziny protoonkogenów, stwierdzono mutacje transformujące jedynie w dwóch kodonach. Najliczniejsze mutacje w genie p53człowieka znajdowano w trzech kodonach CG(N) kodujących argininę: 175, 248 i 273 (ryc. 2). Może to mieć związek ze znaczną podatnością na spontaniczne mutacje dwunukleotydowej sekwencji CpG. W wymienionych trzech kodonach najczęściej dochodzi do tranzycji C do T lub G do A. W wielu przypadkach różnych typów nowotworów, w których stwierdzono mutacje w genie p53, najczęstsze były mutacje zmiany sensu. Insercje (wstawienia zasad) stanowiły 2%, mutacje nonsensowne 6%. Połowa reszty mutacji polegała na zmianie kodonu CGA w TGA i dotyczyła regionu pomiędzy pozycjami 196 a 213 w białkowym produkcie genu. Mutacje dziedziczne genu p53są rzadkie. Na całym świecie stwierdzono ok. 250 takich mutacji. Są one silnie związane z zespołem Li-Fraumeni oraz Li-Fraumeni-podobnym. Występują również w tzw. zespole wielonowotworowym.
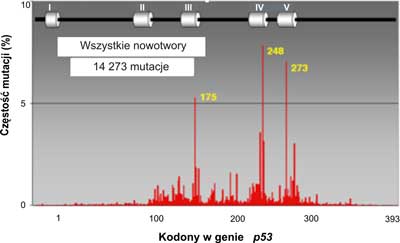
Ryc. 2. Rozrzut mutacji w ludzkim genie p53.
Analiza mutacji wskazuje na pewną współzależność między rodzajem mutacji a narządem, w którym rozwija się nowotwór. Świadczy to o tym, że zachodzi związek przyczynowy między rodzajem zmian mutacyjnych w genie p53a charakterem komórek lub tkanek, w których dochodzi do nowotworzenia oraz czynnikiem uszkadzającym DNA. Jednym z takich czynników jest produkt peroksydacji lipidów, którego głównym najbardziej reaktywnym składnikiem jest trans -4-hydroxy-2-nonenal (HNE). Może on powstawać w wyniku stanów zapalnych w komórkach i reagować bezpośrednio z DNA blokując jego replikacje DNA, tworząc wiązania krzyzowe ( crosslinks) (ryc. 3-5) na obu niciach; transkrybowanej i nietranskrybowanej.
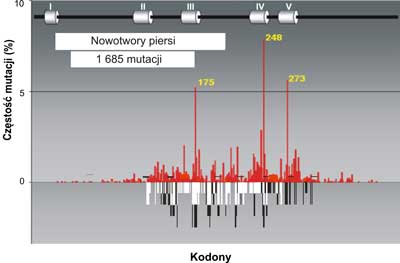
Ryc. 3. Porównanie mutacji w genie p53w nowotworach piersi (szare słupki) do spectrum uszkodzeń indukowanych przez produkt peroksydacji lipidów: trans -4-hydroxy-2-nonenalu (HNE) czarne słupki na nici transkrybowanej i białe słupki na nici nietranskrybowanej egzonach 5-8 p53gene (badania własne). Mutacje w nowotworach piersi wykorzystano z bazy danych (http://perso.curie.fr/Thierry.Soussi/p53_mutation).
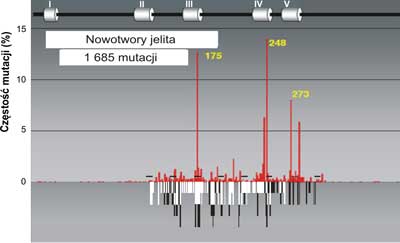
Ryc. 4. Porównanie mutacji w genie p53w nowotworach jelita (szare słupki) do spectrum uszkodzeń indukowanych przez produkt peroksydacji lipidów: trans -4-hydroxy-2-nonenalu (HNE) czarne słupki na nici transkrybowanej i białe słupki na nici nietranskrybowanej egzonach 5-8 p53gene (badania własne). Mutacje w nowotworach jelita wykorzystano z bazy danych (http://perso.curie.fr/Thierry.Soussi/p53_mutation).

Ryc. 5. Porównanie mutacji w genie p53w nowotworach prostaty (szare słupki) do spectrum uszkodzeń indukowanych przez produkt peroksydacji lipidów: trans -4-hydroxy-2-nonenalu (HNE) czarne słupki na nici transkrybowanej i białe słupki na nici nietranskrybowanej egzonach 5-8 p53gene (badania własne). Mutacje w nowotworach prostaty wykorzystano z bazy danych (http://perso.curie.fr/Thierry.Soussi/p53_mutation).
Zauważono, że dość częsta mutacja w obrębie kodonu 175, 248 i 273 spotykana w DNA licznych nowotworów np. piersi, jelita grubego, czy prostaty nigdy nie występuje w DNA raka płuc. Przykłady rozrzutu mutacji dla tych narządów w genie p53człowieka przedstawiają ryciny 3, 4, 5.
Właściwości i rola biologiczna białka p53
Okres połowicznego rozpadu białka p53wynosi około 20 minut. Jego krótka trwałość biologiczna utrudnia prowadzenie badań. Prawidłowe białko p53wykrywane jest w jądrze komórkowym, podczas gdy forma zmutowana – jedynie w cytoplazmie. Funkcjonalną postacią białka p53jest cząsteczka tetrametryczna Sekwencja aminokwasów w pobliżu C – końca cząsteczki białka determinuje możliwość oligomeryzacji i transportu do jądra komórkowego (8). Zasadniczym efektem działania prawidłowego białka p53 jest hamowanie wzrostu i różnicowania hodowanych in vitro komórek myszy i człowieka (9). Białko p53ma zdolność wiąząnia się z białkowymi produktami onkogennych wirusów: wirusa SV40, adenowirusa Eb1, wirusa Papilloma E6, a także z czynnikiem o najwyższej wśród białek aktywności transformacyjnej z produktem genu mdm2. Wykazano że wymienione wyżej białka przyłączają się do cząsteczki białka p53w różnych miejscach (10). Zdolność do wiązania się z białkiem p53wykazują również kinazy białkowe cdc2, kinaza kazeinowa II, a także białka szoku termicznego Hsc 70. Tetramer prawidłowego białka p53może wiązać się z dwuniciowym DNA (1). Charakter wiązania może być zarówno specyficzny, jak i niespecyficzny. Jedno takie miejsce wiązania zidentyfikowano w N – końcowym odcinku białka, w obrębie aminokwasów 1-42, gdzie zgrupowane są aminokwasy kwaśne. W wyniku przebadania wielu genomowych DNA stwierdzono, że prawidłowe białko p53wiąże się z 20-nukleotydowym palindromem występującym w niektórych DNA. Zdolność p53do wiązania się z DNA zanika, gdy białko to jest związane z dużym produktem genu mdm2 – antygenem wirusa SV 40 lub tworzy heterooligomery zawierające w swym składzie zmutowane podjednostki. Wykazano, że białko p53 jeżeli jest w jądrze może działać jako czynnik transkrypcyjny, wiązać się do specyficznych sekwencji DNA, aktywując transkrypcję ponad 60 genów przyległych do miejsca wiązania. Są to białka biorące udział w apoptozie oraz uczestniczące w naprawie DNA lub zatrzymaniu cyklu komórkowego. Natomiast, jeżeli p53 znajdzie się w cytoplazmie – nie ma możliwości regulacji ekspresji genów. Nie jest jednak jasne, gdzie dokładnie w cytoplazmie p53jest przechowywane i w jaki sposób jest transportowany do jądra.
Białko p53wiąże się do DNA, używając wszystkich swoich czterech „ramion”. Typowe miejsce wiązania dla całej molekuły składa się z trzech części: miejsca wiązania specyficznego dla dwóch domen p53różniących się długością od 0 do 13 par zasad i drugiego specyficznego miejsca wiązania dla pozostałych dwóch domen p53, dwie domeny p53są połączone z DNA w pobliżu górnej części łańcucha, a dwie pozostałe w identycznym miejscu wiązania w dole łańcucha. Domena tetrameryzacji ( tetramerization domain) położona jest za helisą, wiążąc wszystkie cztery łańcuchy razem i cztery domeny transaktywacji rozciągające się wzdłuż helisy DNA, gotowa do aktywowania sąsiednich protein zaangażowanych w odczytywanie DNA (ryc. 6). Giętkie łańcuchy łączą się z ze wszystkimi czterema ramionami, co umożliwia p53wiązanie się do wielu różnych rodzajów miejsc w DNA, pozwalając na regulowanie transkrypcji w wielu miejscach w genomie.
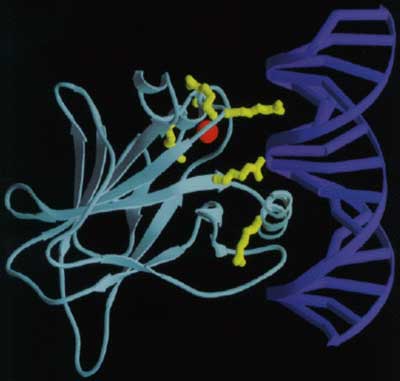
Ryc. 6. Struktura domeny rdzeniowej białka p53w wiązaniu się z helisą DNA. W wiązaniu domeny rdzeniowej p53do DNA kluczową rolę odgrywa sześć aminokwasów oraz atom cynku przedruk z publikacji Cho,Y. et al. (1994) Science, 265, 346-355.
Prawidłowe białko p53 może między innymi indukować apoptozę, podczas gdy białko zmutowane nie wykazuje takich właściwości. W innych przypadkach, stwierdzono efekt odwrotny – hamowanie transkrypcji przez prawidłowe białko p53 (12). Mutacje w obrębie czterech HCD II-IV pozbawiają cząsteczkę tego białka zdolności wpływania na transkrypcję (13). Heterooligomery – kompleksy zmutowanego białka p53z prawidłowym nie wiążą się z DNA i nie wywierają wpływu na transkrypcję. Zauważono, że w hodowli prawidłowych komórek myszy, poziom białka p53zmienia się w poszczególnych fazach cyklu komórkowego, osiągając w fazie G1 najwyższe wartości. W innych doświadczeniach, zauważono że wniknięcie białka p53do jądra komórkowego powoduje zatrzymanie cyklu komórkowego w późnej fazie G1 (14). Rola białka p53 w cyklu komórkowym staje się szczególnie widoczna w warunkach stresu. Na przykład, w wyniku uszkodzenia DNA promieniowaniem jonizującym, produkcja białka p53w komórkach ulega szybkiemu zwiększeniu. Jest to dla komórki korzystne, gdyż pozostawia jej czas na naprawę uszkodzonego materiału genetycznego. W niektórych komórkach, w których doszło do poważnego uszkodzenia DNA, które nie może być szybko naprawione, obserwuje się charakterystyczne zmiany (fragmentację jądra). Przyjmuje się, że dochodzi wtedy do indukcji apoptozy – programowanej śmierci komórki. Stężenie białka p53w komórce musi być jednak ściśle regulowane, gdyż zbyt duża jego ilość mogłaby przyspieszać proces starzenia poprzez zbyt nadmierną apoptozę. Głównym regulatorem poziomu stężenia białka p53jest ligaza Mdm2/Hdm2 (mouse double minute-2/human double minute-2), która poprzez ubikwitynację prowadzi do degradacji p53w proteasomie. Regulacja ta odbywa się na zasadzie ujemnego sprzężenia zwrotnego, gdyż transkrypcja genu mdm2jest aktywowana przez p53. By nie degradować p53w sytuacji, gdy jest ono potrzebne do likwidowania skutków uszkodzenia DNA, ochronę przed ubikwitynacją stanowi fosforylacja przez kinazę cdc2, której maksymalny poziom osiągany jest podczas fazy M.
Białko p53 a transformacja
Gen p53był początkowo uważany za onkogen, ponieważ wydawało się, że jego produkt wywołuje transformację nowotworową. Dopiero w ostatnich latach okazało się, że jedynie zmutowane formy białka p53wykazują aktywność transformacyjną. Zmutowane białko p53wzmaga aktywność transformacyjną genu ras, podczas gdy prawidłowe białko wydatnie hamuje kotransformacje prowadzone jednocześnie przez formy zmutowane białka oraz przez gen ras (15). Aktywność transformacyjna zmutowanych cząsteczek białka p53zależy od rodzaju zmian, do jakich w nich doszło na skutek mutacji. Wykazano, że białko p53zmutowane w kodonie 175 wykazuje od 3 do 10 razy większą aktywność transformacyjną niż gdy mutacja dotyczyła kodonu 273 (16). Wiadomo już, że gen p53odgrywa główną rolę w ochronie organizmu przed rakiem. Komórki rakowe zazwyczaj zawierają dwa typy mutacji: mutacje, które powodują niekontrolowany wzrost i podziały komórek i inne mutacje, które blokują systemy obronne i zabezpieczenie przed nienaturalnym wzrostem. Przeważnie zmiany w takich białkach są bardzo szerokie np. delecja fragmentu genu lub mutacja typu zmiana ramki odczytu ( frame shift). Białko takie jest niezdolne do pełnienia jakiejkolwiek funkcji. Zmiana informacji DNA dotyczy tylko jednej pozycji i powoduje, że komórka buduje p53z błędem, wstawiając niepoprawny aminokwas do łańcucha polipeptydowego. W genie p53mutacje te są nieco inne niż w przypadku pozostałych białek zmutowanych w komórkach rakowych. Są to mutacje punktowe i polegają na substytucji kilku par zasad. Wynikiem tych mutacji jest białko, które ma zmienione najważniejsze aminokwasy odpowiadające za wiązanie DNA. Tetramery, w skład których wchodzi chociaż jeden zmutowany polipeptyd p53są niezdolne do wiązania DNA. W tych mutantach, normalne funkcjonowanie p53jest zablokowane i białko nie może zahamować mnożenia się uszkodzonych komórek. Jeśli komórki posiadają także inne mutacje, może powodować to niekontrolowany wzrost i z komórki prawdopodobnie rozwinie się guz.
Gen p53a kancerogeneza
Doświadczenia ostatnich pięciu lat wykazały, że produkt genu p53 hamuje nowotworzenie. Dlatego powszechnie przyjmuje się, że gen p53 należy zaliczyć do supresorów transformacji nowotworowej. W ponad połowie nowotworów u ludzi stwierdza się mutacje w p53. Analiza mutacji wskazuje na pewną współzależność między rodzajem mutacji a narządem, w którym rozwija się nowotwór (7). Świadczy to o tym, że zachodzi związek przyczynowy między rodzajem zmian mutacyjnych w genie p53a charakterem komórek lub tkanek, w których dochodzi do nowotworzenia oraz czynnikiem uszkadzającym DNA. Zauważono, że dość częsta mutacja w obrębie kodonu 175 spotykana w DNA licznych nowotworów np. pęcherza, jelita grubego, sutka i wątroby, nigdy nie występuje w DNA raka płuc (ryc. 7). W przypadku raka wątroby u osób żyjących na obszarach, w których za czynnik ryzyka uważa się aflatoksynę B1 oraz wirusa B zapalenia wątroby, większość mutacji znajdowano w kodonie 249 (3). Przeprowadzając analizę materiału (obejmującego przypadki), w których różnym typom nowotworów towarzyszyły mutacje w genie p53, zauważono że w DNA komórek jelit, mózgu i układu limfatycznego przeważały tranzycje G:C(r)A:T, podczas gdy w DNA komórek raka płuc i wątroby częstsze były transwersje, szczególnie G:C (r) T:A. (ryc. 7). Wskaźnik transwersji wyraźnie zależał od typu nowotworu. W DNA komórek raka wątroby transwersje obejmowały 20% badanych przypadków, zaś w DNA komórek raka płuc 40%.
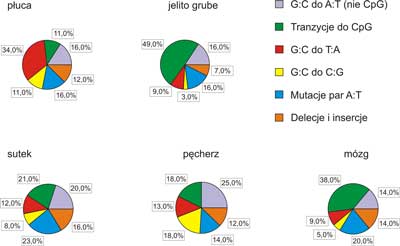
Ryc. 7. Procentowy udział typów mutacji w nowotworach różnych organów.
Prawdopodobnie istnieje związek pomiędzy dużą liczbą mutacji znajdowanych w komórkach raka płuc a podatnością guaniny na atak zewnętrznych czynników chemicznych (17).
Najlepiej przebadano mutacje genu p53w komórkach raka jelita grubego. W około 80% przypadków stwierdzono anomalie w obrębie obydwu alleli: delecje i mutacje punktowe. W około 75% przypadków obszar delecji znajdowano w chromosomie 17p (18). Fakty te mogą wskazywać, że rak jelita grubego rozwija się przede wszystkim wtedy gdy brak jest prawidłowego produktu genu p53. Wykazano, że komórki produkujące zmutowane formy białka p53częściej ulegają transformacji nowotworowej niż komórki pozbawione tego białka (19).
Molekularny mechanizm działania białka p53
Wysunięto dwie hipotezy tłumaczące molekularny mechanizm działania białka p53. Na podstawie wyników doświadczeń, w których badano wpływ białka na syntezę DNA (14). Sformułowano pierwszą hipotezę, która przyjmuje że funkcja biologiczna białka p53polega na hamowaniu replikacji DNA na etapie inicjacji (12). Przypuszcza się, że prawidłowe białko p53wiąże się z jeszcze niezidentyfikowanym białkiem replikacyjnym, co powoduje zablokowanie syntezy DNA i uniemożliwia wejście w fazę S. Prawdopodobnie istnieje komórkowy odpowiednik antygenu T, który bierze udział w inicjacji replikacji DNA. Związanie się białka p53z tym komórkowym odpowiednikiem wirusowego antygenu uniemożliwia replikację. Według drugiej hipotezy, zasadniczą funkcją białka p53jest regulacja procesu transkrypcji. Zakłada się, że może ono działać jako transaktywator transkrypcji stymulując lub hamując syntezę m RNA. Białko p53może aktywować transkrypcję genów przyczyniających się do hamowania wzrostu komórki lub powodować represję genów działających przeciwnie. Ostatecznym efektem działania białka p53byłoby więc hamowanie wzrostu komórki. Drugą interesującą koncepcję roli biologicznej produktu genu p53przedstawił Lane (20). Według tego autora, w prawidłowo funkcjonującej komórce białko p53pełni rolę „strażnika genomu”. Gdy DNA ulega uszkodzeniu wskutek oddziaływania czynników pochodzenia zewnętrznego lub wewnętrznego, białko p53nagromadza się i wyłącza replikację, aby zapewnić czas na naprawę uszkodzeń. Gdy reperacja nie dochodzi do skutku, białko p53przyczynia się do uruchomienia procesu apoptozy (zaprogramowanej śmierci) przy współudziale innych białek, co pozwala na wyeliminowanie nieprawidłowej komórki ze środowiska (ryc. 8). Proces apoptozy pozwala na utrzymanie homeostazy w organizmie. W komórkach, w których białko p53zostało zinaktywowane przez mutacje lub zostało związane z białkami blokującymi jego funkcje, uszkodzony DNA ulega replikacji. Nie dochodzi do zatrzymania cyklu komórkowego w fazie G1, a komórki ulegają przyspieszonemu wzrostowi i podziałom. W zbyt szybko dzielących się komórkach dochodzi do częstych mutacji i rearanżacji chromosomowych, co doprowadza do szybkiej selekcji uzłośliwionych klonów. Komórki rakowe skupiają się na niszczeniu p53 tak, aby uzyskać nieśmiertelność. Dlatego właściwości białka p53 są rozpatrywane jako jeden z celów przyszłej strategii leczenia raka. Terapia, która zmuszałaby komórki rakowe do popełnienia autodestrukcji, mogłaby zatrzymywać rozwój prawie wszystkich typów raka. Dlatego też, produkt genu p53 można uważać za ważny czynnik regulujący wzrost, proliferację i różnicowanie komórek.
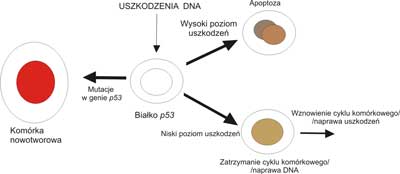
Ryc. 8. Sugerowany model działania białka p53jako „strażnika genomu”.
Poznanie dodatkowych i nowych funkcji genu p53, może pozwolić na przewidywanie wpływu określonych mutacji również na funkcję jego białka, a w konsekwencji na lepszą profilaktykę w chorobach nowotworowych. Mimo 20 lat badań nad genem p53, nasza wiedza na ten temat jest nadal niekompletna, a wiele pytań pozostaje nadal bez odpowiedzi.
Piśmiennictwo
1. Vogelstein B et al.: A deadly inheritance. Nature (Lond) 1990; 348: 681-682. 2. Oren M et al.: Tumor spressor teaming up to restrain cancer. EMBO J 1992; 3: 2179-2183. 3. Hollstein M et al.: p53Mutations in Human Cancers. Sciense, 1991; Vol. 253: 49-53. 4. Wolf D et al.: In vitro expression of human p53cDNA clones and characterization of the cloned human p53gene. Molecular Cell Biology 1985; 5: 1887-1893. 5. Lamb P et al.: p53, the cellular gatekeeper for growth and division. Mol Cel Biol 1986; 6: 1379-1385. 6. Matlasevski GJ et al.: Telling changes of base. Mol Cel Biol 1987; 7: 961-963. 7. Caron de Fromentel et al.: Rainbow trout p53cDNA doning and biochemical characterization. Genes Chromosomes and Cancer 1992; 4: 1-15. 8. Sidransky D et al.: Identyfication of ras Oncogene Mutations in the Stool of Patients with Curable Colorectal Tumors. Science 1992; 256: 102-105. 9. Chen PL et al.: Genetic mechanisms of Tumor Supression by the Human gene p53. Science 1990; 250: 1576-1580. 10. Barak Y et al.: p53tumor supressor gene of model for investigating human mutagenesis. EMBO J 1992; 11: 2115-2121. 11. Cho Y et al.: Crystal structure of a p53tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 1994; 265: 346-355. 12. Gannon JV, Lane DP: p53and DNA polimerase a compete for binding to SV 40 T antigen. Nature 1987; 329: 456-458. 13. Farmer G et al.: Wild-type p53activates transcription in vitro. Nature 1992; 358: 83-86. 14. Kastan MB et al.: Participation of p53protein in the cellular response to DNA damage. Cancer Reseach 1991; 51: 6034-6311. 15. Zambetti GP et al.: A mutant p53protein is required for maintenance of the transformed phenotype in cells transformed with p53plus ras cDNAs. Proc Natl Acad Sci USA 1992; 89: 3952-3956. 16. Hinds PW et al.: Distribution of DNA damage in chromatin and its relation and its relation to repair in human cells. Mol Cel Biol 1987; 7: 2863-2869. 17. Kriek E et al.: Carcionogenesis by aromatic amines. Biochim Biophys Acta 1984; 738: 181-201. 18. Baker SJ et al.: p53gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Reseach 1990; 50: 7717-7722. 19. Harris CC et al.: p53: at the crossroads of molecular carcinogenenesis and molecular epidemiology. N Eng J Med 1993; 329: 1318-1327. 20. Lane DP et al.: p53, gurdian of the genome. Nature 1992; 316: 15-16.
otrzymano/received: 2010-03-03
zaakceptowano/accepted: 2010-04-07
Adres/address:
*Paweł Kowalczyk
Interdyscyplinarne Centrum Modelowania Matematycznego i Komputerowego UW
ul. Pawińskiego 5a, 02-106 Warszawa
tel.: (22) 592 22 44
e-mail: pawelk@ibb.waw.pl